Expected output
InStrain produces a variety of output in the IS folder depending on which operations are run. Generally, output that is meant for human eyes to be easily interpretable is located in the output folder.
inStrain profile
A typical run of inStrain will yield the following files in the output folder:
scaffold_info.tsv
This gives basic information about the scaffolds in your sample at the highest allowed level of read identity.
scaffold |
length |
coverage |
breadth |
nucl_diversity |
coverage_median |
coverage_std |
coverage_SEM |
breadth_minCov |
breadth_expected |
nucl_diversity_median |
nucl_diversity_rarefied |
nucl_diversity_rarefied_median |
breadth_rarefied |
conANI_reference |
popANI_reference |
SNS_count |
SNV_count |
divergent_site_count |
consensus_divergent_sites |
population_divergent_sites |
N5_271_010G1_scaffold_100 |
1148 |
1.89808362369338 |
0.9764808362369338 |
0.0 |
2 |
1.0372318863390368 |
0.030626273060932862 |
0.018292682926829267 |
0.8128805020451009 |
0.0 |
0.0 |
1.0 |
1.0 |
0 |
0 |
0 |
0 |
0 |
||
N5_271_010G1_scaffold_102 |
1144 |
2.388986013986014 |
0.9956293706293706 |
0.003678160837326971 |
2 |
1.3042095721915248 |
0.038576628450898466 |
0.07604895104895107 |
0.8786983245100435 |
0.0 |
0.0 |
1.0 |
1.0 |
0 |
0 |
0 |
0 |
0 |
||
N5_271_010G1_scaffold_101 |
1148 |
1.7439024390243902 |
0.9599303135888502 |
2 |
0.8728918441975071 |
0.025773816178570358 |
0.0 |
0.7855901382035807 |
0.0 |
0.0 |
0.0 |
0 |
00 |
0 |
0 |
|||||
N5_271_010G1_scaffold_103 |
1142 |
2.039404553415061 |
0.9938704028021016 |
0.0 |
2 |
1.1288397384374758 |
0.03341869350286944 |
0.04028021015 |
- scaffold
The name of the scaffold in the input .fasta file
- length
Full length of the scaffold in the input .fasta file
- coverage
The average depth of coverage on the scaffold. If half the bases in a scaffold have 5 reads on them, and the other half have 10 reads, the coverage of the scaffold will be 7.5
- breadth
The percentage of bases in the scaffold that are covered by at least a single read. A breadth of 1 means that all bases in the scaffold have at least one read covering them
- nucl_diversity
The mean nucleotide diversity of all bases in the scaffold that have a nucleotide diversity value calculated. So if only 1 base on the scaffold meets the minimum coverage to calculate nucleotide diversity, the nucl_diversity of the scaffold will be the nucleotide diversity of that base. Will be blank if no positions have a base over the minimum coverage.
- coverage_median
The median depth of coverage value of all bases in the scaffold, included bases with 0 coverage
- coverage_std
The standard deviation of all coverage values
- coverage_SEM
The standard error of the mean of all coverage values (calculated using scipy.stats.sem)
- breadth_minCov
The percentage of bases in the scaffold that have at least min_cov coverage (e.g. the percentage of bases that have a nucl_diversity value and meet the minimum sequencing depth to call SNVs)
- breadth_expected
expected breadth; this tells you the breadth that you should expect if reads are evenly distributed along the genome, given the reported coverage value. Based on the function breadth = -1.000 * e^(0.883 * coverage) + 1.000. This is useful to establish whether or not the scaffold is actually in the reads, or just a fraction of the scaffold. If your coverage is 10x, the expected breadth will be ~1. If your actual breadth is significantly lower then the expected breadth, this means that reads are mapping only to a specific region of your scaffold (transposon, prophage, etc.)
- nucl_diversity_median
The median nucleotide diversity value of all bases in the scaffold that have a nucleotide diversity value calculated
- nucl_diversity_rarefied
The average nucleotide diversity among positions that have at least
--rarefied_coverage(50x by default). These values are also calculated by randomly subsetting the reads at that position to--rarefied_coveragereads- nucl_diversity_rarefied_median
The median rarefied nucleotide diversity (similar to that described above)
- breadth_rarefied
The percentage of bases in a scaffold that have at least
--rarefied_coverage- conANI_reference
The conANI between the reads and the reference genome
- popANI_reference
The popANI between the reads and the reference genome
- SNS_count
The total number of SNSs called on this scaffold
- SNV_count
The total number of SNVs called on this scaffold
- divergent_site_count
The total number of divergent sites called on this scaffold
- consensus_divergent_sites
The total number of divergent sites in which the reads have a different consensus allele than the reference genome. These count as “differences” in the conANI_reference calculation, and
breadth_minCov*lengthcounts as the denominator.- population_divergent_sites
The total number of divergent sites in which the reads do not have the reference genome base as any allele at all (major or minor). These count as “differences” in the popANI_reference calculation, and
breadth_minCov*lengthcounts as the denominator.
mapping_info.tsv
This provides an overview of the number of reads that map to each scaffold, and some basic metrics about their quality. The header line (starting with #; not shown in the table below) describes the parameters that were used to filter the reads
scaffold |
pass_pairing_filter |
filtered_pairs |
median_insert |
mean_PID |
pass_min_insert |
unfiltered_reads |
unfiltered_pairs |
pass_min_read_ani |
filtered_priority_reads |
unfiltered_singletons |
mean_insert_distance |
pass_min_mapq |
mean_mistmaches |
mean_mapq_score |
unfiltered_priority_reads |
pass_max_insert |
filtered_singletons |
mean_pair_length |
all_scaffolds |
22886 |
9435 |
318.75998426985933 |
0.942328296264744 |
22804.0 |
71399 |
22886 |
9499.0 |
0 |
25627 |
322.1849602376999 |
22886.0 |
14.325963471117715 |
17.16896792799091 |
0 |
22828.0 |
0 |
255.52 |
N5_271_010G1_scaffold_1 |
432 |
346 |
373.0 |
0.9719013034762376 |
432.0 |
959 |
432 |
346.0 |
0 |
95 |
373.72222222222223 |
432.0 |
7.643518518518517 |
33.030092592592595 |
0 |
432.0 |
0 |
274.7106481481 |
N5_271_010G1_scaffold_0 |
741 |
460 |
389.0 |
0.9643004762700924 |
740.0 |
1841 |
741 |
461.0 |
0 |
359 |
387.94466936572195 |
741.0 |
10.2361673414305 |
26.537112010796218 |
0 |
741.0 |
0 |
285.5033738191 |
N5_271_010G1_scaffold_2 |
348 |
252 |
369.5 |
0.965446218901576 |
347.0 |
865 |
348 |
253.0 |
0 |
169 |
349.0172413793104 |
348.0 |
8.227011494252874 |
31.557471264367813 |
0 |
347.0 |
0 |
243.3103448275 |
N5_271_010G1_scaffold_3 |
301 |
205 |
367.0 |
0.9639376512009891 |
301.0 |
1088 |
301 |
205.0 |
0 |
486 |
327.81395348837214 |
301.0 |
8.70764119601329 |
29.089700996677745 |
0 |
300.0 |
0 |
251.2624584717 |
N5_271_010G1_scaffold_4 |
213 |
153 |
389.0 |
0.9649427929020106 |
213.0 |
502 |
213 |
153.0 |
0 |
76 |
372.3896713615024 |
213.0 |
9.27699530516432 |
30.70422535211268 |
0 |
213.0 |
0 |
269.2300469483 |
N5_271_010G1_scaffold_5 |
134 |
114 |
366.0 |
0.977820509122326 |
134.0 |
349 |
134 |
116.0 |
0 |
81 |
376.4552238805969 |
134.0 |
5.164179104477612 |
37.61194029850746 |
0 |
132.0 |
0 |
246.8059701492 |
N5_271_010G1_scaffold_6 |
140 |
130 |
384.5 |
0.9813174696928879 |
140.0 |
316 |
140 |
130.0 |
0 |
36 |
372.45 |
140.0 |
4.864285714285714 |
38.43571428571428 |
0 |
140.0 |
0 |
261.3071428571429 |
- scaffold
The name of the scaffold in the input .fasta file. For the top row this will read
all_scaffolds, and it has the sum of all rows.- pass_pairing_filter
The number of individual reads that pass the selecting pairing filter (only paired reads will pass this filter by default)
- filtered_pairs
The number of pairs of reads that pass all cutoffs
- median_insert
Among all pairs of reads mapping to this scaffold, the median insert distance.
- mean_PID
Among all pairs of reads mapping to this scaffold, the average percentage ID of both reads in the pair to the reference .fasta file
- pass_min_insert
The number of pairs of reads mapping to this scaffold that pass the minimum insert size cutoff
- unfiltered_reads
The raw number of reads that map to this scaffold
- unfiltered_pairs
The raw number of pairs of reads that map to this scaffold. Only paired reads are used by inStrain
- pass_min_read_ani
The number of pairs of reads mapping to this scaffold that pass the min_read_ani cutoff
- filtered_priority_reads
The number of priority reads that pass the rest of the filters (will only be non-0 if a priority reads input file is provided)
- unfiltered_singletons
The number of reads detected in which only one read of the pair is mapped
- mean_insert_distance
Among all pairs of reads mapping to this scaffold, the mean insert distance. Note that the insert size is measured from the start of the first read to the end of the second read (2 perfectly overlapping 50bp reads will have an insert size of 50bp)
- pass_min_mapq
The number of pairs of reads mapping to this scaffold that pass the minimum mapQ score cutoff
- mean_mistmaches
Among all pairs of reads mapping to this scaffold, the mean number of mismatches
- mean_mapq_score
Among all pairs of reads mapping to this scaffold, the average mapQ score
- unfiltered_priority_reads
The number of reads that pass the pairing filter because they were part of the
priority_readsinput file (will only be non-0 if a priority reads input file is provided).- pass_max_insert
The number of pairs of reads mapping to this scaffold that pass the maximum insert size cutoff- that is, their insert size is less than 3x the median insert size of all_scaffolds. Note that the insert size is measured from the start of the first read to the end of the second read (2 perfectly overlapping 50bp reads will have an insert size of 50bp)
- filtered_singletons
The number of reads detected in which only one read of the pair is mapped AND which make it through to be considered. This will only be non-0 if the filtering settings allows non-paired reads
- mean_pair_length
Among all pairs of reads mapping to this scaffold, the average length of both reads in the pair summed together
Warning
Adjusting the pairing filter will result in odd values for the “filtered_pairs” column; this column reports the number of pairs AND singletons that pass the filters. To calculate the true number of filtered pairs, use the formula filtered_pairs - filtered_singletons
SNVs.tsv
This describes the SNVs and SNSs that are detected in this mapping. While we should refer to these mutations as divergent sites, sometimes SNV is used to refer to both SNVs and SNSs
Warning
inStrain reports 0-based values for “position”. The first base in a scaffold will be position “0”, second based position “1”, etc.
scaffold |
position |
position_coverage |
allele_count |
ref_base |
con_base |
var_base |
ref_freq |
con_freq |
var_freq |
A |
C |
T |
G |
gene |
mutation |
mutation_type |
cryptic |
class |
N5_271_010G1_scaffold_120 |
174 |
5 |
2 |
C |
C |
A |
0.6 |
0.6 |
0.4 |
2 |
3 |
0 |
0 |
I |
False |
SNV |
||
N5_271_010G1_scaffold_120 |
195 |
6 |
1 |
T |
C |
A |
0.0 |
1.0 |
0.0 |
0 |
6 |
0 |
0 |
I |
False |
SNS |
||
N5_271_010G1_scaffold_120 |
411 |
8 |
2 |
A |
A |
C |
0.75 |
0.75 |
0.25 |
6 |
2 |
0 |
0 |
N5_271_010G1_scaffold_120_1 |
N:V163G |
N |
False |
SNV |
N5_271_010G1_scaffold_120 |
426 |
9 |
2 |
G |
G |
T |
0.7777777777777778 |
0.7777777777777778 |
0.2222222222222222 |
0 |
0 |
2 |
7 |
N5_271_010G1_scaffold_120_1 |
N:S178Y |
N |
False |
SNV |
N5_271_010G1_scaffold_120 |
481 |
6 |
2 |
C |
T |
C |
0.3333333333333333 |
0.6666666666666666 |
0.3333333333333333 |
0 |
2 |
4 |
0 |
N5_271_010G1_scaffold_120_1 |
N:D233N |
N |
False |
con_SNV |
N5_271_010G1_scaffold_120 |
484 |
6 |
2 |
G |
A |
G |
0.3333333333333333 |
0.6666666666666666 |
0.3333333333333333 |
4 |
0 |
0 |
2 |
N5_271_010G1_scaffold_120_1 |
N:P236S |
N |
False |
con_SNV |
N5_271_010G1_scaffold_120 |
488 |
5 |
1 |
T |
C |
T |
0.2 |
0.8 |
0.2 |
0 |
4 |
1 |
0 |
N5_271_010G1_scaffold_120_1 |
S:240 |
S |
False |
SNS |
N5_271_010G1_scaffold_120 |
811 |
5 |
1 |
T |
A |
T |
0.2 |
0.8 |
0.2 |
4 |
0 |
1 |
0 |
N5_271_010G1_scaffold_120_1 |
N:N563Y |
N |
False |
SNS |
N5_271_010G1_scaffold_120 |
897 |
7 |
2 |
G |
G |
T |
0.7142857142857143 |
0.7142857142857143 |
0.2857142857142857 |
0 |
0 |
2 |
5 |
I |
False |
SNV |
See the module_descriptions for what constitutes a SNP (what makes it into this table)
- scaffold
The scaffold that the SNV is on
- position
The genomic position of the SNV
- position_coverage
The number of reads detected at this position
- allele_count
The number of bases that are detected above background levels (according to the null model. An allele_count of 0 means no bases are supported by the reads, an allele_count of 1 means that only 1 base is supported by the reads, an allele_count of 2 means two bases are supported by the reads, etc.
- ref_base
The reference base in the .fasta file at that position
- con_base
The consensus base (the base that is supported by the most reads)
- var_base
Variant base; the base with the second most reads
- ref_freq
The fraction of reads supporting the ref_base
- con_freq
The fraction of reds supporting the con_base
- var_freq
The fraction of reads supporting the var_base
- A, C, T, and G
The number of mapped reads encoding each of the bases
- gene
If a gene file was included, this column will be present listing if the SNV is in the coding sequence of a gene
- mutation
Short-hand code for the amino acid switch. If synonymous, this will be S: + the position. If nonsynonymous, this will be N: + the old amino acid + the position + the new amino acid. NOTE - the position of the amino acid is always calculated from left to right on the genome file, whether or not it’s the forward or reverse strand. Codons are calculated correctly (considering strandedness), this only impacts the reported “position” in this column. I know this is weird behavior and it will change in future inStrain versions.
- mutation_type
What type of mutation this is. N = nonsynonymous, S = synonymous, I = intergenic, M = there are multiple genes with this base so you cant tell
- cryptic
If an SNV is cryptic, it means that it is detected when using a lower read mismatch threshold, but becomes undetected when you move to a higher read mismatch level. See “dealing with mm” in the advanced_use section for more details on what this means.
- class
The classification of this divergent site. The options are SNS (meaning allele_count is 1 and con_base does not equal ref_base), SNV (meaning allele_count is > 1 and con_base does equal ref_base), con_SNV (meaning allele_count is > 1, con_base does not equal ref_base, and ref_base is present in the reads; these count as differences in conANI calculations), pop_SNV (meaning allele_count is > 1, con_base does not equal ref_base, and ref_base is not present in the reads; these count as differences in popANI and conANI calculations), DivergentSite (meaning allele count is 0), and AmbiguousReference (meaning the ref_base is not A, C, T, or G)
linkage.tsv
This describes the linkage between pairs of SNPs in the mapping that are found on the same read pair at least min_snp times.
Warning
inStrain reports 0-based values for “position”. The first base in a scaffold will be position “0”, second based position “1”, etc.
scaffold |
position_A |
position_B |
distance |
r2 |
d_prime |
r2_normalized |
d_prime_normalized |
allele_A |
allele_a |
allele_B |
allele_b |
countab |
countAb |
countaB |
countAB |
total |
N5_271_010G1_scaffold_93 |
58 |
59 |
1 |
0.021739130434782702 |
1.0 |
0.031141868512110725 |
1.0 |
C |
T |
G |
A |
0 |
3 |
4 |
20 |
27 |
N5_271_010G1_scaffold_93 |
58 |
70 |
12 |
0.012820512820512851 |
1.0 |
C |
T |
T |
A |
0 |
2 |
4 |
22 |
28 |
||
N5_271_010G1_scaffold_93 |
58 |
80 |
22 |
0.016722408026755814 |
1.0 |
0.005847953216374271 |
1.0 |
C |
T |
G |
A |
0 |
2 |
5 |
21 |
28 |
N5_271_010G1_scaffold_93 |
58 |
84 |
26 |
0.7652173913043475 |
1.0000000000000002 |
0.6296296296296297 |
1.0 |
C |
T |
G |
C |
4 |
0 |
1 |
22 |
27 |
N5_271_010G1_scaffold_93 |
58 |
101 |
43 |
0.00907029478458067 |
1.0 |
C |
T |
C |
A |
0 |
2 |
2 |
19 |
23 |
||
N5_271_010G1_scaffold_93 |
58 |
126 |
68 |
0.01754385964912257 |
1.0 |
0.002770083102493075 |
1.0 |
C |
T |
A |
T |
0 |
2 |
3 |
16 |
21 |
N5_271_010G1_scaffold_93 |
58 |
133 |
75 |
0.008333333333333352 |
1.0 |
C |
T |
G |
T |
0 |
1 |
3 |
17 |
21 |
||
N5_271_010G1_scaffold_93 |
59 |
70 |
11 |
0.010869565217391413 |
1.0 |
0.02777777777777779 |
1.0 |
G |
A |
T |
A |
0 |
2 |
3 |
21 |
26 |
N5_271_010G1_scaffold_93 |
59 |
80 |
21 |
0.6410256410256397 |
1.0 |
1.0 |
1.0 |
G |
A |
G |
A |
2 |
0 |
1 |
25 |
28 |
Linkage is used primarily to determine if organisms are undergoing horizontal gene transfer or not. It’s calculated for pairs of SNPs that can be connected by at least min_snp reads. It’s based on the assumption that each SNP has two alleles (for example, a A and b B). This all gets a bit confusing and has a large amount of literature around each of these terms, but I’ll do my best to briefly explain what’s going on
- scaffold
The scaffold that both SNPs are on
- position_A
The position of the first SNP on this scaffold
- position_B
The position of the second SNP on this scaffold
- distance
The distance between the two SNPs
- r2
This is the r-squared linkage metric. See below for how it’s calculated
- d_prime
This is the d-prime linkage metric. See below for how it’s calculated
- r2_normalized, d_prime_normalized
These are calculated by rarefying to
min_snpnumber of read pairs. See below for how it’s calculated- allele_A
One of the two bases at position_A
- allele_a
The other of the two bases at position_A
- allele_B
One of the bases at position_B
- allele_b
The other of the two bases at position_B
- countab
The number of read-pairs that have allele_a and allele_b
- countAb
The number of read-pairs that have allele_A and allele_b
- countaB
The number of read-pairs that have allele_a and allele_B
- countAB
The number of read-pairs that have allele_A and allele_B
- total
The total number of read-pairs that have have information for both position_A and position_B
Python code for the calculation of these metrics:
freq_AB = float(countAB) / total
freq_Ab = float(countAb) / total
freq_aB = float(countaB) / total
freq_ab = float(countab) / total
freq_A = freq_AB + freq_Ab
freq_a = freq_ab + freq_aB
freq_B = freq_AB + freq_aB
freq_b = freq_ab + freq_Ab
linkD = freq_AB - freq_A * freq_B
if freq_a == 0 or freq_A == 0 or freq_B == 0 or freq_b == 0:
r2 = np.nan
else:
r2 = linkD*linkD / (freq_A * freq_a * freq_B * freq_b)
linkd = freq_ab - freq_a * freq_b
# calc D-prime
d_prime = np.nan
if (linkd < 0):
denom = max([(-freq_A*freq_B),(-freq_a*freq_b)])
d_prime = linkd / denom
elif (linkD > 0):
denom = min([(freq_A*freq_b), (freq_a*freq_B)])
d_prime = linkd / denom
################
# calc rarefied
rareify = np.random.choice(['AB','Ab','aB','ab'], replace=True, p=[freq_AB,freq_Ab,freq_aB,freq_ab], size=min_snp)
freq_AB = float(collections.Counter(rareify)['AB']) / min_snp
freq_Ab = float(collections.Counter(rareify)['Ab']) / min_snp
freq_aB = float(collections.Counter(rareify)['aB']) / min_snp
freq_ab = float(collections.Counter(rareify)['ab']) / min_snp
freq_A = freq_AB + freq_Ab
freq_a = freq_ab + freq_aB
freq_B = freq_AB + freq_aB
freq_b = freq_ab + freq_Ab
linkd_norm = freq_ab - freq_a * freq_b
if freq_a == 0 or freq_A == 0 or freq_B == 0 or freq_b == 0:
r2_normalized = np.nan
else:
r2_normalized = linkd_norm*linkd_norm / (freq_A * freq_a * freq_B * freq_b)
# calc D-prime
d_prime_normalized = np.nan
if (linkd_norm < 0):
denom = max([(-freq_A*freq_B),(-freq_a*freq_b)])
d_prime_normalized = linkd_norm / denom
elif (linkd_norm > 0):
denom = min([(freq_A*freq_b), (freq_a*freq_B)])
d_prime_normalized = linkd_norm / denom
rt_dict = {}
for att in ['r2', 'd_prime', 'r2_normalized', 'd_prime_normalized', 'total', 'countAB', \
'countAb', 'countaB', 'countab', 'allele_A', 'allele_a', \
'allele_B', 'allele_b']:
rt_dict[att] = eval(att)
gene_info.tsv
This describes some basic information about the genes being profiled
Warning
inStrain reports 0-based values for “position”, including the “start” and “stop” in this table. The first base in a scaffold will be position “0”, second based position “1”, etc.
scaffold |
gene |
gene_length |
coverage |
breadth |
breadth_minCov |
nucl_diversity |
start |
end |
direction |
partial |
dNdS_substitutions |
pNpS_variants |
SNV_count |
SNV_S_count |
SNV_N_count |
SNS_count |
SNS_S_count |
SNS_N_count |
divergent_site_count |
N5_271_010G1_scaffold_0 |
N5_271_010G1_scaffold_0_1 |
141.0 |
0.7092198581560284 |
0.7092198581560284 |
0.0 |
143 |
283 |
-1 |
False |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
|||
N5_271_010G1_scaffold_0 |
N5_271_010G1_scaffold_0_2 |
219.0 |
4.849315068493151 |
1.0 |
0.45662100456620996 |
0.012312216758728069 |
2410 |
2628 |
-1 |
False |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
||
N5_271_010G1_scaffold_0 |
N5_271_010G1_scaffold_0_3 |
282.0 |
7.528368794326241 |
1.0 |
0.9609929078014184 |
0.00805835530326815 |
3688 |
3969 |
-1 |
False |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
||
N5_271_010G1_scaffold_1 |
N5_271_010G1_scaffold_1_1 |
336.0 |
2.7261904761904763 |
1.0 |
0.0625 |
0.0 |
0 |
335 |
-1 |
False |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
||
N5_271_010G1_scaffold_1 |
N5_271_010G1_scaffold_1_2 |
717.0 |
7.714086471408647 |
1.0 |
0.8926080892608089 |
0.011336830817162968 |
378 |
1094 |
-1 |
False |
0.554203539823008 |
9.0 |
2.0 |
6.0 |
0.0 |
0.0 |
0.0 |
9.0 |
|
N5_271_010G1_scaffold_1 |
N5_271_010G1_scaffold_1_3 |
114.0 |
13.105263157894735 |
1.0 |
1.0 |
0.016291986431991808 |
1051 |
1164 |
-1 |
False |
0.3956834532374099 |
4.0 |
1.0 |
2.0 |
0.0 |
0.0 |
0.0 |
4.0 |
|
N5_271_010G1_scaffold_1 |
N5_271_010G1_scaffold_1_4 |
111.0 |
11.342342342342342 |
1.0 |
1.0 |
0.02102806761458109 |
1164 |
1274 |
-1 |
False |
5.0 |
0.0 |
5.0 |
0.0 |
0.0 |
0.0 |
5.0 |
||
N5_271_010G1_scaffold_1 |
N5_271_010G1_scaffold_1_5 |
174.0 |
9.057471264367816 |
1.0 |
1.0 |
0.006896087493019509 |
1476 |
1649 |
-1 |
False |
0.0 |
2.0 |
2.0 |
0.0 |
0.0 |
0.0 |
0.0 |
2.0 |
|
N5_271_010G1_scaffold_1 |
N5_271_010G1_scaffold_1_6 |
174.0 |
6.195402298850576 |
1.0 |
0.7413793103448276 |
0.028698649055273976 |
1656 |
1829 |
-1 |
False |
0.5790697674418601 |
4.0 |
1.0 |
3.0 |
0.0 |
0.0 |
0.0 |
4.0 |
- scaffold
Scaffold that the gene is on
- gene
Name of the gene being profiled
- gene_length
Length of the gene in nucleotides
- breadth
The number of bases in the gene that have at least 1x coverage
- breadth_minCov
The number of bases in the gene that have at least min_cov coverage
- nucl_diversity
The mean nucleotide diversity of all bases in the gene that have a nucleotide diversity value calculated. So if only 1 base on the scaffold meets the minimum coverage to calculate nucleotide diversity, the nucl_diversity of the scaffold will be the nucleotide diversity of that base. Will be blank if no positions have a base over the minimum coverage.
- start
Start of the gene (position on scaffold; 0-indexed)
- end
End of the gene (position on scaffold; 0-indexed)
- direction
Direction of the gene (based on prodigal call). If -1, means the gene is not coded in the direction expressed by the .fasta file
- partial
If True this is a partial gene; based on not having partial=00 in the record description provided by Prodigal
- dNdS_substitutions
The dN/dS of SNSs detected in this gene. Will be blank if 0 N and/or 0 S substitutions are detected
- pNpS_variants
The pN/pS of SNVs detected in this gene. Will be blank if 0 N and/or 0 S SNVs are detected
- SNV_count
Total number of SNVs detected in this gene
- SNV_S_count
Number of synonymous SNVs detected in this gene
- SNV_N_count
Number of non-synonymous SNVs detected in this gene
- SNS_count
Total number of SNSs detected in this gens
- SNS_S_count
Number of synonymous SNSs detected in this gens
- SNS_N_count
Number of non-synonymous SNSs detected in this gens
- divergent_site_count
Number of divergent sites detected in this gens
genome_info.tsv
Describes many of the above metrics on a genome-by-genome level, rather than a scaffold-by-scaffold level.
genome |
coverage |
breadth |
nucl_diversity |
length |
true_scaffolds |
detected_scaffolds |
coverage_median |
coverage_std |
coverage_SEM |
breadth_minCov |
breadth_expected |
nucl_diversity_rarefied |
conANI_reference |
popANI_reference |
iRep |
iRep_GC_corrected |
linked_SNV_count |
SNV_distance_mean |
r2_mean |
d_prime_mean |
consensus_divergent_sites |
population_divergent_sites |
SNS_count |
SNV_count |
filtered_read_pair_cou |
nt |
reads_unfiltered_pairs |
reads_mean_PID |
reads_unfiltered_reads |
divergent_site_count |
|||||||||||||||||||||
fobin.fasta |
132.07770270270268 |
0.9974662162162162 |
0.035799449026225894 |
1184 |
1 |
1 |
113 |
114.96590198492832 |
3.6668428018497408 |
0.9822635135135136 |
1.0 |
0.034319907739082 |
0.979363714531 |
||||||||||||
3844 |
0.9939810834049873 |
False |
1064.0 |
120.48214285714286 |
0.07781470898619759 |
0.8710788695476385 |
24 |
7 |
7 |
97 |
926 |
5991 |
0.9239440924157436 |
19260 |
104 |
||||||||||
maxbin2.maxbin.001.fasta |
6.5637243038012985 |
0.8940915760335204 |
0.007116301715134402 |
264436 |
166 |
166 |
5 |
9.475490303923918 |
0.019704930458769948 |
0.5080246259964604 |
0.99695960719657 |
0.0002 |
|||||||||||||
8497234066195295 |
0.997201131457496 |
0.9990248622897128 |
False |
777.0 |
80.73101673101674 |
0.2979679685064011 |
0.9518999449773424 |
376 |
131 |
127 |
1246 |
7368 |
9309 |
0.9783316024248924 |
2 |
||||||||||
5281 |
1373 |
- genome
The name of the genome being profiled. If all scaffolds were a single genome, this will read “all_scaffolds”
- coverage
Average coverage depth of all scaffolds of this genome
- breadth
The breadth of all scaffolds of this genome
- nucl_diversity
The average nucleotide diversity of all scaffolds of this genome
- length
The full length of this genome across all scaffolds
- true_scaffolds
The number of scaffolds present in this genome based off of the scaffold-to-bin file
- detected_scaffolds
The number of scaffolds with at least a single read-pair mapping to them
- coverage_median
The median coverage among all bases in the genome
- coverage_std
The standard deviation of all coverage values
- coverage_SEM
The standard error of the mean of all coverage values (calculated using scipy.stats.sem)
- breadth_minCov
The percentage of bases in the scaffold that have at least min_cov coverage (e.g. the percentage of bases that have a nucl_diversity value and meet the minimum sequencing depth to call SNVs)
- breadth_expected
This tells you the breadth that you should expect if reads are evenly distributed along the genome, given the reported coverage value. Based on the function breadth = -1.000 * e^(0.883 * coverage) + 1.000. This is useful to establish whether or not the scaffold is actually in the reads, or just a fraction of the scaffold. If your coverage is 10x, the expected breadth will be ~1. If your actual breadth is significantly lower then the expected breadth, this means that reads are mapping only to a specific region of your scaffold (transposon, prophage, etc.)
- nucl_diversity_rarefied
The average nucleotide diversity among positions that have at least
--rarefied_coverage(50x by default). These values are also calculated by randomly subsetting the reads at that position to--rarefied_coveragereads- conANI_reference
The conANI between the reads and the reference genome
- popANI_reference
The popANI between the reads and the reference genome
- iRep
The iRep value for this genome (if it could be successfully calculated)
- iRep_GC_corrected
A True / False value of whether the iRep value was corrected for GC bias
- linked_SNV_count
The number of divergent sites that could be linked in this genome
- SNV_distance_mean
Average distance between linked divergent sites
- r2_mean
Average r2 between linked SNVs (see explanation of linkage.tsv above for more info)
- d_prime_mean
Average d prime between linked SNVs (see explanation of linkage.tsv above for more info)
- consensus_divergent_sites
The total number of divergent sites in which the reads have a different consensus allele than the reference genome. These count as “differences” in the conANI_reference calculation, and
breadth_minCov*lengthcounts as the denominator.- population_divergent_sites
The total number of divergent sites in which the reads do not have the reference genome base as any allele at all (major or minor). These count as “differences” in the popANI_reference calculation, and
breadth_minCov*lengthcounts as the denominator.- SNS_count
The total number of SNSs called on this genome
- SNV_count
The total number of SNVs called on this genome
- filtered_read_pair_count
The total number of read pairs that pass filtering and map to this genome
- reads_unfiltered_pairs
The total number of pairs, filtered or unfiltered, that map to this genome
- reads_mean_PID
The average ANI of mapped read pairs to the reference genome for this genome
- reads_unfiltered_reads
The total number of reads, filtered or unfiltered, that map to this genome
- divergent_site_count
The total number of divergent sites called on this genome
inStrain parse_annotations
A typical run of inStrain parse_gene_annotations will yield the following files in the output folder. For more information, see User Manual
LongFormData.csv
All of the annotation information a very long table
sample |
anno |
genomes |
genes |
bases |
2bag10_1.bam |
K03737 |
{‘REFINED_METABAT215_TOP10_CONTIGS_1500_ASSEMBLY_K77_MERGED__Hadza_MoBio_hadza-E-H_A_23_1707.16.fa’} |
1 |
6666 |
2bag10_1.bam |
K06973 |
{‘REFINED_METABAT215_TOP10_CONTIGS_1500_ASSEMBLY_K77_MERGED__Hadza_MoBio_hadza-E-H_A_23_1707.16.fa’} |
1 |
1068 |
2bag10_1.bam |
K04066 |
{‘REFINED_METABAT215_TOP10_CONTIGS_1500_ASSEMBLY_K77_MERGED__Hadza_MoBio_hadza-E-H_A_23_1707.16.fa’, ‘Bifidobacterium_longum_subsp_infantis_ATCC_15697.fna’} |
2 |
195761 |
2bag10_1.bam |
K15558 |
{‘REFINED_METABAT215_TOP10_CONTIGS_1500_ASSEMBLY_K77_MERGED__Hadza_MoBio_hadza-E-H_A_23_1707.16.fa’, ‘Bifidobacterium_longum_subsp_infantis_ATCC_15697.fna’} |
96 |
10748749 |
2bag10_1.bam |
K19762 |
{‘REFINED_METABAT215_TOP10_CONTIGS_1500_ASSEMBLY_K77_MERGED__Hadza_MoBio_hadza-E-H_A_23_1707.16.fa’, ‘Bifidobacterium_longum_subsp_infantis_ATCC_15697.fna’} |
97 |
10920075 |
2bag10_1.bam |
3000025 |
{‘REFINED_METABAT215_TOP10_CONTIGS_1500_ASSEMBLY_K77_MERGED__Hadza_MoBio_hadza-E-H_A_23_1707.16.fa’, ‘Bifidobacterium_longum_subsp_infantis_ATCC_15697.fna’} |
2 |
168916 |
2bag10_1.bam |
K18888 |
{‘REFINED_METABAT215_TOP10_CONTIGS_1500_ASSEMBLY_K77_MERGED__Hadza_MoBio_hadza-E-H_A_23_1707.16.fa’, ‘Bifidobacterium_longum_subsp_infantis_ATCC_15697.fna’} |
3 |
504008 |
2bag10_1.bam |
K20386 |
{‘REFINED_METABAT215_TOP10_CONTIGS_1500_ASSEMBLY_K77_MERGED__Hadza_MoBio_hadza-E-H_A_23_1707.16.fa’, ‘Bifidobacterium_longum_subsp_infantis_ATCC_15697.fna’} |
98 |
11007871 |
2bag10_1.bam |
K07979 |
{‘REFINED_METABAT215_TOP10_CONTIGS_1500_ASSEMBLY_K77_MERGED__Hadza_MoBio_hadza-E-H_A_23_1707.16.fa’} |
1 |
742 |
- sample
The sample this row refers to (based on the name of the .bam file used to create the inStrain profile)
- anno
The annotation this row refers to (based on the input annotation table)
- genomes
The specific genomes that have this particular annotation. Represented as a python set
- genes
The total number of genes detected with this annotation in this sample
- bases
The total number of base-pairs mapped to all genes with this annotation in this sample
SampleAnnotationTotals.csv
Totals for each sample. Used to generate the _fraction tables enumerated below.
sample |
detected_genes |
detected_genomes |
bases_mapped_to_genes |
detected_annotations |
detected_genes_with_anno |
2bag10_1.bam |
2625 |
2 |
222405987 |
3302 |
1677 |
2bag10_2.bam |
20909 |
10 |
2418511040 |
32225 |
15513 |
- sample
The sample this row refers to (based on the name of the .bam file used to create the inStrain profile)
- detected_genes
The total number of genes detected in this sample after passing the set filters
- detected_genomes
The total number of genomes detected in this sample after passing the set filters
- bases_mapped_to_genes
The total number of bases mapped to detected genes. See ParsedGeneAnno_bases.csv below for more info
- detected_annotations
The total number of annotations detected; this can be higher than detected_genes_with_anno if some genes have multiple annotations
- detected_genes_with_anno
The total number of genes detected with at least one annotation
ParsedGeneAnno_*.csv
There are a total of 6 tables like this generated in the output folder, each looking like the following:
sample |
3000005 |
3000024 |
3000025 |
3000026 |
3000027 |
3000074 |
3000118 |
3000165 |
3000166 |
2bag10_1.bam |
131097 |
1286827 |
168916 |
1656 |
0 |
0 |
0 |
0 |
0 |
2bag10_2.bam |
104013 |
5016854 |
955645 |
2552 |
633275 |
1034042 |
95617 |
409295 |
541951 |
In each case the column sample is the sample the row refers to (based on the name of the .bam file used to create the inStrain profile), and all other columns are annotations from the input annotation_table provides. The number values differ depending on the individual output table being analyzed. Below you can find descriptions on what the numbers refer to:
- ParsedGeneAnno_bases.csv
The total number of base pairs mapped to all genes with this annotation. The number of base pairs mapped for each gene with this annotation is calculated as the gene length * the coverage of the gene, and the number reported is the sum of this value of all genes
- ParsedGeneAnno_bases_fraction.csv
The values in ParsedGeneAnno_bases.csv divided by the total number of bases mapped to all detected genes (the value bases_mapped_to_genes reported in SampleAnnotationTotals.csv)
- ParsedGeneAnno_genes.csv
The total number of detected genes with this annotation
- ParsedGeneAnno_genes_fraction.csv
The values in ParsedGeneAnno_genes.csv divided by the total number of genes detected (the value detected_genes reported in SampleAnnotationTotals.csv)
- ParsedGeneAnno_genomes.csv
The total number of genomes with at least one detected gene with this annotation
- ParsedGeneAnno_genomes_fraction.csv
The values in ParsedGeneAnno_genomes.csv divided by the total number of genomes detected (the value detected_genomes reported in SampleAnnotationTotals.csv)
inStrain compare
A typical run of inStrain will yield the following files in the output folder:
comparisonsTable.tsv
Summarizes the differences between two inStrain profiles on a scaffold by scaffold level
scaffold |
name1 |
name2 |
coverage_overlap |
compared_bases_count |
percent_genome_compared |
length |
consensus_SNPs |
population_SNPs |
popANI |
conANI |
N5_271_010G1_scaffold_98 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
1.0 |
61 |
0.05290546400693842 |
1153 |
0 |
0 |
1.0 |
1.0 |
N5_271_010G1_scaffold_133 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
1.0 |
78 |
0.0741444866920152 |
1052 |
0 |
0 |
1.0 |
1.0 |
N5_271_010G1_scaffold_144 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
1.0 |
172 |
0.16715257531584066 |
1029 |
0 |
0 |
1.0 |
1.0 |
N5_271_010G1_scaffold_158 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
1.0 |
36 |
0.035749751737835164 |
1007 |
0 |
0 |
1.0 |
1.0 |
N5_271_010G1_scaffold_57 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
1.0 |
24 |
0.0183206106870229 |
1310 |
0 |
0 |
1.0 |
1.0 |
N5_271_010G1_scaffold_139 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
1.0 |
24 |
0.023121387283236997 |
1038 |
0 |
0 |
1.0 |
1.0 |
N5_271_010G1_scaffold_92 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
1.0 |
336 |
0.286934244235696 |
1171 |
0 |
0 |
1.0 |
1.0 |
N5_271_010G1_scaffold_97 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
1.0 |
22 |
0.01901469317199654 |
1157 |
0 |
0 |
1.0 |
1.0 |
N5_271_010G1_scaffold_100 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
1.0 |
21 |
0.018292682926829267 |
1148 |
0 |
0 |
1.0 |
1.0 |
- scaffold
The scaffold being compared
- name1
The name of the first inStrain profile being compared
- name2
The name of the second inStrain profile being compared
- coverage_overlap
The percentage of bases that are either covered or not covered in both of the profiles (covered = the base is present at at least min_snp coverage). The formula is length(coveredInBoth) / length(coveredInEither). If both scaffolds have 0 coverage, this will be 0.
- compared_bases_count
The number of considered bases; that is, the number of bases with at least min_snp coverage in both profiles. Formula is length([x for x in overlap if x == True]).
- percent_genome_compared
The percentage of bases in the scaffolds that are covered by both. The formula is length([x for x in overlap if x == True])/length(overlap). When ANI is np.nan, this must be 0. If both scaffolds have 0 coverage, this will be 0.
- length
The total length of the scaffold
- consensus_SNPs
The number of locations along the genome where both samples have the base at >= 5x coverage, and the consensus allele in each sample is different. Used to calculate conANI
- population_SNPs
The number of locations along the genome where both samples have the base at >= 5x coverage, and no alleles are shared between either sample. Used to calculate popANI
- popANI
The average nucleotide identity among compared bases between the two scaffolds, based on population_SNPs. Calculated using the formula popANI = (compared_bases_count - population_SNPs) / compared_bases_count
- conNI
The average nucleotide identity among compared bases between the two scaffolds, based on consensus_SNPs. Calculated using the formula conANI = (compared_bases_count - consensus_SNPs) / compared_bases_count
pairwise_SNP_locations.tsv
Warning
inStrain reports 0-based values for “position”. The first base in a scaffold will be position “0”, second based position “1”, etc.
Lists the locations of all differences between profiles. Because it’s a big file, this will only be created is you include the flag --store_mismatch_locations in your inStrain compare command.
scaffold |
position |
name1 |
name2 |
consensus_SNP |
population_SNP |
con_base_1 |
ref_base_1 |
var_base_1 |
position_coverage_1 |
A_1 |
C_1 |
T_1 |
G_1 |
con_base_2 |
ref_base_2 |
var_base_2 |
position_coverage_2 |
A_2 |
C_2 |
T_2 |
G_2 |
N5_271_010G1_scaffold_9 |
823 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
True |
False |
G |
G |
A |
10.0 |
3.0 |
0.0 |
0.0 |
7.0 |
A |
G |
G |
6.0 |
3.0 |
0.0 |
0.0 |
3.0 |
N5_271_010G1_scaffold_11 |
906 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
True |
False |
T |
T |
C |
6.0 |
0.0 |
2.0 |
4.0 |
0.0 |
C |
T |
T |
7.0 |
0.0 |
4.0 |
3.0 |
0.0 |
N5_271_010G1_scaffold_29 |
436 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
True |
False |
C |
T |
T |
6.0 |
0.0 |
3.0 |
3.0 |
0.0 |
T |
T |
C |
7.0 |
0.0 |
3.0 |
4.0 |
0.0 |
N5_271_010G1_scaffold_140 |
194 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
True |
False |
A |
A |
T |
6.0 |
4.0 |
0.0 |
2.0 |
0.0 |
T |
A |
A |
9.0 |
4.0 |
0.0 |
5.0 |
0.0 |
N5_271_010G1_scaffold_24 |
1608 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
True |
False |
G |
G |
A |
8.0 |
2.0 |
0.0 |
0.0 |
6.0 |
A |
G |
G |
6.0 |
5.0 |
0.0 |
0.0 |
1.0 |
N5_271_010G1_scaffold_112 |
600 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
True |
False |
A |
G |
G |
6.0 |
4.0 |
0.0 |
0.0 |
2.0 |
||||||||
N5_271_010G1_scaffold_88 |
497 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
True |
False |
A |
G |
G |
5.0 |
3.0 |
0.0 |
0.0 |
2.0 |
||||||||
N5_271_010G1_scaffold_53 |
1108 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
True |
False |
A |
A |
G |
5.0 |
3.0 |
0.0 |
0.0 |
2.0 |
G |
A |
A |
15.0 |
6.0 |
0.0 |
0.0 |
9.0 |
N5_271_010G1_scaffold_46 |
710 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
True |
False |
A |
C |
C |
6.0 |
4.0 |
2.0 |
0.0 |
0.0 |
C |
C |
A |
6.0 |
2.0 |
4.0 |
0.0 |
0.0 |
- scaffold
The scaffold on which the difference is located
- position
The position where the difference is located (0-based)
- name1
The name of the first inStrain profile being compared
- name2
The name of the second inStrain profile being compared
- consensus_SNP
A True / False column listing whether or not this difference counts towards conANI calculations
- population_SNP
A True / False column listing whether or not this difference counts towards popANI calculations
- con_base_1
The consensus base of the profile listed in
name1at this position- ref_base_1
The reference base of the profile listed in
name1at this position (will be the same asref_base_2)- var_base_1
The variant base of the profile listed in
name1at this position- position_coverage_1
The number of reads mapping to this position in
name1- A_1, C_1, T_1, G_1
The number of mapped reads with each nucleotide in
name1- con_base_2, ref_base_2, …
The above columns are also listed for the
name2sample
genomeWide_compare.tsv
A genome-level summary of the differences detected by inStrain compare. Generated by running inStrain genome_wide on the results of inStrain compare, or by providing an stb file to the original inStrain compare command.
genome |
name1 |
name2 |
coverage_overlap |
compared_bases_count |
consensus_SNPs |
population_SNPs |
popANI |
conANI |
percent_compared |
all_scaffolds |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
1.0 |
100712 |
0 |
0 |
1.0 |
1.0 |
0.3605549091560011 |
all_scaffolds |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
0.6852932198159855 |
71900 |
196 |
50.9999304589707928 |
0.9972739916550765 |
0.25740624720307886 |
|
all_scaffolds |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
1.0 |
145663 |
0 |
0 |
1.0 |
1.0 |
0.5214821444553835 |
- genome
The genome being compared
- name1
The name of the first inStrain profile being compared
- name2
The name of the second inStrain profile being compared
- coverage_overlap
The percentage of bases that are either covered or not covered in both of the profiles (covered = the base is present at at least min_snp coverage). The formula is length(coveredInBoth) / length(coveredInEither). If both scaffolds have 0 coverage, this will be 0.
- compared_bases_count
The number of considered bases; that is, the number of bases with at least min_snp coverage in both profiles. Formula is length([x for x in overlap if x == True]).
- percent_genome_compared
The percentage of bases in the scaffolds that are covered by both. The formula is length([x for x in overlap if x == True])/length(overlap). When ANI is np.nan, this must be 0. If both scaffolds have 0 coverage, this will be 0.
- length
The total length of the genome
- consensus_SNPs
The number of locations along the genome where both samples have the base at >= 5x coverage, and the consensus allele in each sample is different. Used to calculate conANI
- population_SNPs
The number of locations along the genome where both samples have the base at >= 5x coverage, and no alleles are shared between either sample. Used to calculate popANI
- popANI
The average nucleotide identity among compared bases between the two scaffolds, based on population_SNPs. Calculated using the formula popANI = (compared_bases_count - population_SNPs) / compared_bases_count
- conNI
The average nucleotide identity among compared bases between the two scaffolds, based on consensus_SNPs. Calculated using the formula conANI = (compared_bases_count - consensus_SNPs) / compared_bases_count
strain_clusters.tsv
The result of clustering the pairwise comparison data provided in genomeWide_compare.tsv to generate strain-level clusters. Performed using hierarchical clustering in the same manner as the program dRep; see the dRep documentation for some info on the oddities of hierarchical clustering
cluster |
sample |
genome |
1_1 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
fobin.fasta |
1_1 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
fobin.fasta |
2_1 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
maxbin2.maxbin.001.fasta |
2_2 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
maxbin2.maxbin.001.fasta |
- cluster
The strain identity of this genome in this sample. Each “strain” assigned by the hierarchical clustering algorithm will have a unique cluster. In the example table above strains of the genome
fobin.fastaare the same in both samples (they have the same “cluster” identities), but strains of the genomemaxbin2.maxbin.001.fastaare different in the two samples (they have different “cluster” identities).- sample
The sample that the genome was detected in.
- genome
The genome that the cluster is referring to.
pooled_SNV_info.tsv, pooled_SNV_data.tsv, and pooled_SNV_data_keys.tsv
The tables pooled_SNV_info.tsv, pooled_SNV_data.tsv, and pooled_SNV_data_keys.tsv can be generated by inStrain compare by providing .bam files to the inStrain compare command. See User Manual for more information.
scaffold |
position |
depth |
A |
C |
T |
G |
ref_base |
con_base |
var_base |
sample_detections |
sample_5x_detections |
DivergentSite_count |
SNS_count |
SNV_count |
con_SNV_count |
pop_SNV_count |
sample_con_bases |
N5_271_010G1_scaffold_114 |
3 |
10 |
2 |
0 |
7 |
1 |
T |
T |
A |
2 |
1 |
0 |
0 |
1 |
0 |
0 |
[‘T’] |
N5_271_010G1_scaffold_114 |
20 |
33 |
0 |
31 |
2 |
0 |
C |
C |
T |
2 |
2 |
0 |
0 |
1 |
0 |
0 |
[‘C’] |
N5_271_010G1_scaffold_114 |
24 |
35 |
29 |
0 |
2 |
4 |
A |
A |
G |
2 |
2 |
0 |
0 |
2 |
0 |
0 |
[‘A’] |
N5_271_010G1_scaffold_114 |
25 |
38 |
2 |
36 |
0 |
0 |
C |
C |
A |
2 |
2 |
0 |
0 |
1 |
0 |
0 |
[‘C’] |
N5_271_010G1_scaffold_114 |
55 |
71 |
66 |
5 |
0 |
0 |
A |
A |
C |
2 |
2 |
0 |
0 |
1 |
0 |
0 |
[‘A’] |
N5_271_010G1_scaffold_114 |
57 |
67 |
2 |
0 |
0 |
65 |
G |
G |
A |
2 |
2 |
0 |
0 |
1 |
0 |
0 |
[‘G’] |
N5_271_010G1_scaffold_114 |
75 |
95 |
4 |
90 |
0 |
1 |
C |
C |
A |
2 |
2 |
0 |
0 |
1 |
0 |
0 |
[‘C’] |
N5_271_010G1_scaffold_114 |
76 |
95 |
0 |
90 |
2 |
3 |
C |
C |
G |
2 |
2 |
0 |
0 |
2 |
0 |
0 |
[‘C’] |
N5_271_010G1_scaffold_114 |
79 |
98 |
0 |
3 |
0 |
95 |
G |
G |
C |
2 |
2 |
0 |
0 |
1 |
0 |
0 |
[‘G’] |
This table has information about each SNV, summarized across all samples
- scaffold
The scaffold being analyzed
- position
The position in the scaffold where the SNV is located (0-based)
- depth
The total number of reads mapping to this scaffold across samples
- A
The number of reads with A at this position in this scaffold across samples
- C
The number of reads with C at this position in this scaffold across samples
- T
The number of reads with T at this position in this scaffold across samples
- G
The number of reads with G at this position in this scaffold across samples
- ref_base
The reference base at this position in this scaffold across samples
- con_base
The consensus base (most common) at this position in this scaffold across samples
- var_base
The variant base (second most common) at this position in this scaffold across samples
- sample_detections
The number of samples in which this position at this scaffold has at least one read mapping to it
- sample_5x_detections
The number of samples in which this position at this scaffold has at least 5 reads mapping to it
- DivergentSite_count
The number of samples with a divergent sites detected at this position
- SNS_count
The number of samples with a SNSs detected at this position
- SNV_count
The number of samples with a SNVs detected at this position
- con_SNV_count
The number of samples with consenus SNPs (conANI) detected at this position
- pop_SNV_count
The number of samples with population SNPs (popANI) detected at this position
- sample_con_bases
The number of different consensus bases at this position across all analyzed samples
sample |
scaffold |
position |
A |
C |
T |
G |
0 |
0 |
3 |
2 |
0 |
5 |
1 |
0 |
0 |
20 |
0 |
21 |
2 |
0 |
0 |
0 |
24 |
21 |
0 |
0 |
4 |
0 |
0 |
25 |
2 |
26 |
0 |
0 |
0 |
0 |
55 |
38 |
5 |
0 |
0 |
0 |
0 |
57 |
2 |
0 |
0 |
38 |
0 |
0 |
75 |
3 |
55 |
0 |
0 |
0 |
0 |
76 |
0 |
56 |
0 |
3 |
0 |
0 |
79 |
0 |
1 |
0 |
57 |
This table has information about each SNV in each sample. Because the table can be huge, names of scaffolds and samples are listed as “keys” to be translated using the also-provided pooled_SNV_data_keys.tsv table
- sample
The key for the sample being analyzed (as detailed in the pooled_SNV_data_keys.tsv table below)
- scaffold
The key for the scaffold being analyzed (as detailed in the pooled_SNV_data_keys.tsv table below)
- position
The position in the scaffold where the SNV is located (0-based)
- A,C,T,G
The number of reads with this base in this sample in this scaffold at this position
key |
sample |
scaffold |
0 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G1.sorted.bam |
N5_271_010G1_scaffold_114 |
1 |
N5_271_010G1_scaffold_min1000.fa-vs-N5_271_010G2.sorted.bam |
N5_271_010G1_scaffold_63 |
2 |
N5_271_010G1_scaffold_89 |
|
3 |
N5_271_010G1_scaffold_33 |
|
4 |
N5_271_010G1_scaffold_95 |
|
5 |
N5_271_010G1_scaffold_11 |
|
6 |
N5_271_010G1_scaffold_74 |
|
7 |
N5_271_010G1_scaffold_71 |
|
8 |
N5_271_010G1_scaffold_96 |
This table has “keys” needed to translate the pooled_SNV_data.tsv table
- key
The key in question. This is the number presented in the sample or scaffold column in the pooled_SNV_data.tsv table above
- sample
The name of the sample with this key. For example: for the row with a 0 as the key, sample 0 in pooled_SNV_data.tsv refers to the sample listed here
- scaffold
The name of the scaffold with this key. For example: for the row with a 0 as the key, scaffold 0 in pooled_SNV_data.tsv refers to the sample listed here
inStrain plot
This is what the results of inStrain plot look like.
1) Coverage and breadth vs. read mismatches
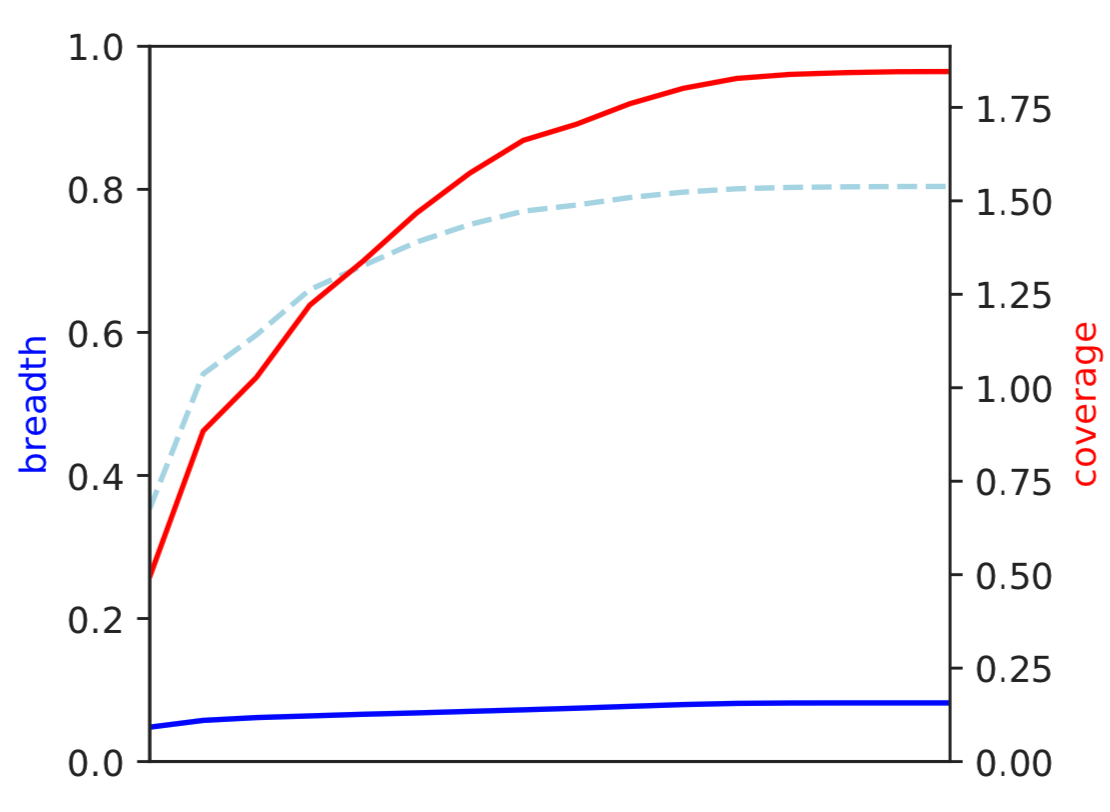
Breadth of coverage (blue line), coverage depth (red line), and expected breadth of coverage given the depth of coverage (dotted blue line) versus the minimum ANI of mapped reads. Coverage depth continues to increase while breadth of plateaus, suggesting that all regions of the reference genome are not present in the reads being mapped.
2) Genome-wide microdiversity metrics
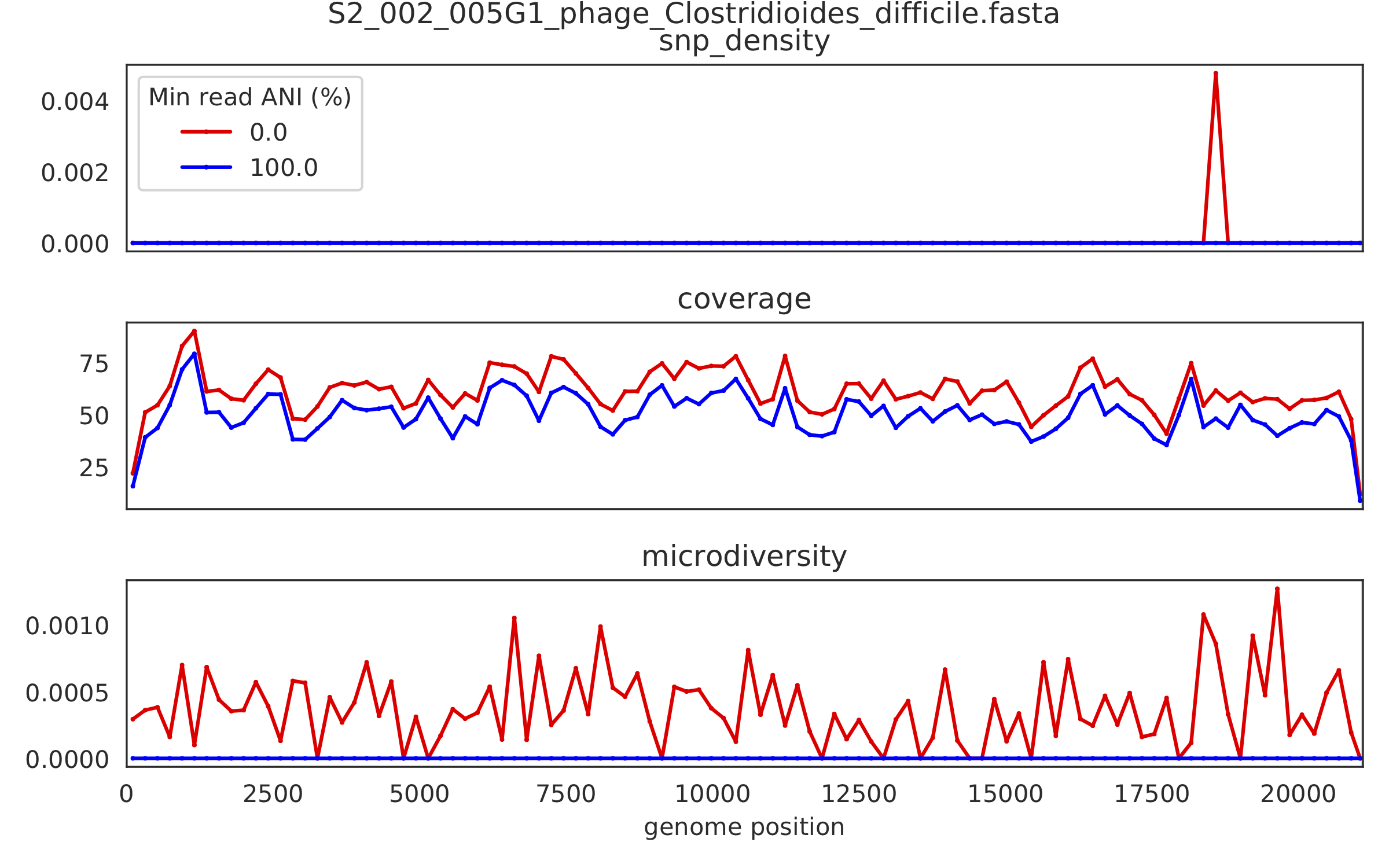
SNV density, coverage, and nucleotide diversity. Spikes in nucleotide diversity and SNV density do not correspond with increased coverage, indicating that the signals are not due to read mis-mapping. Positions with nucleotide diversity and no SNV-density are those where diversity exists but is not high enough to call a SNV
3) Read-level ANI distribution
Distribution of read pair ANI levels when mapped to a reference genome; this plot suggests that the reference genome is >1% different than the mapped reads
4) Major allele frequencies

Distribution of the major allele frequencies of bi-allelic SNVs (the Site Frequency Spectrum). Alleles with major frequencies below 50% are the result of multiallelic sites. The lack of distinct puncta suggest that more than a few distinct strains are present.
5) Linkage decay
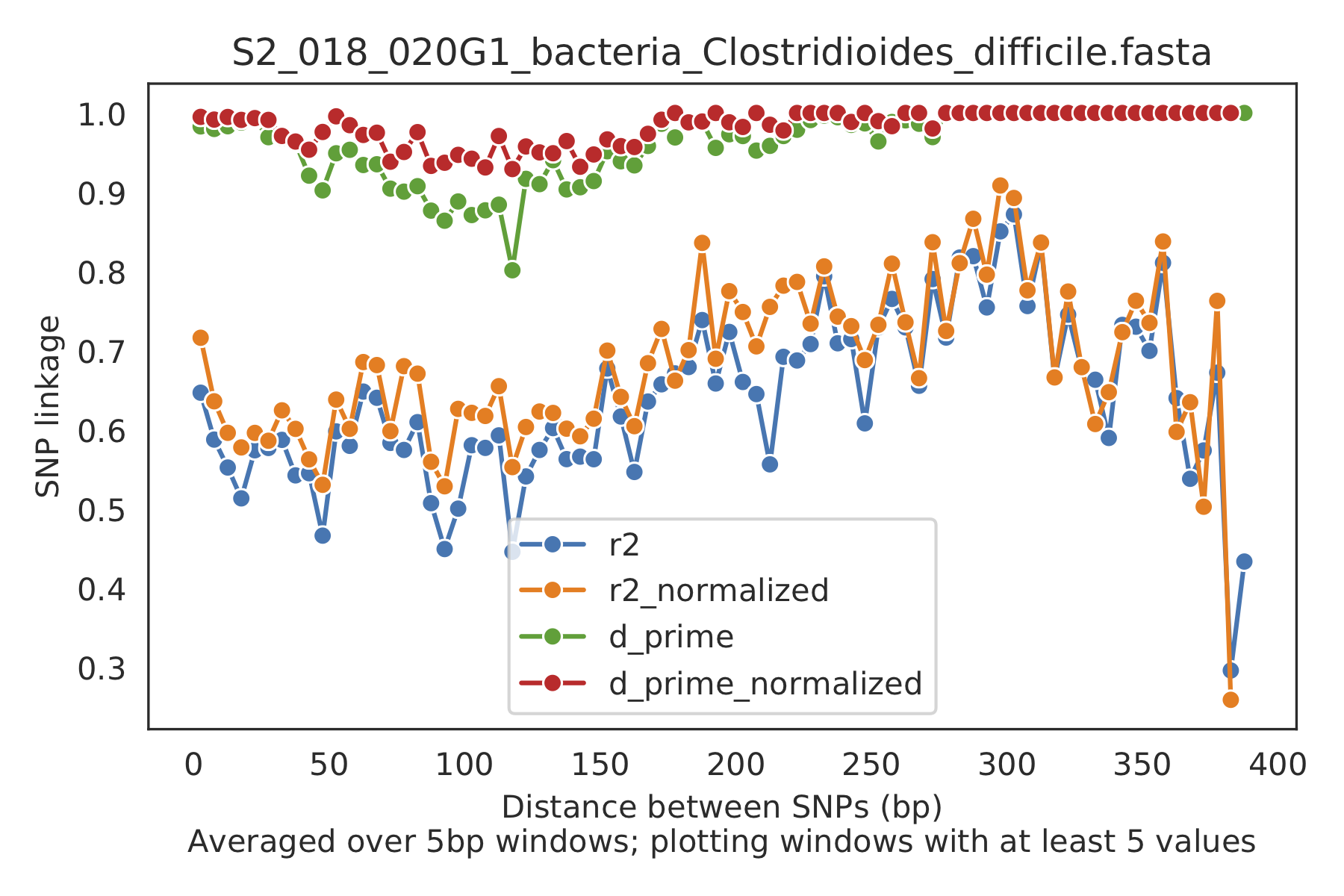

Metrics of SNV linkage vs. distance between SNVs; linkage decay (shown in one plot and not the other) is a common signal of recombination.
6) Read filtering plots
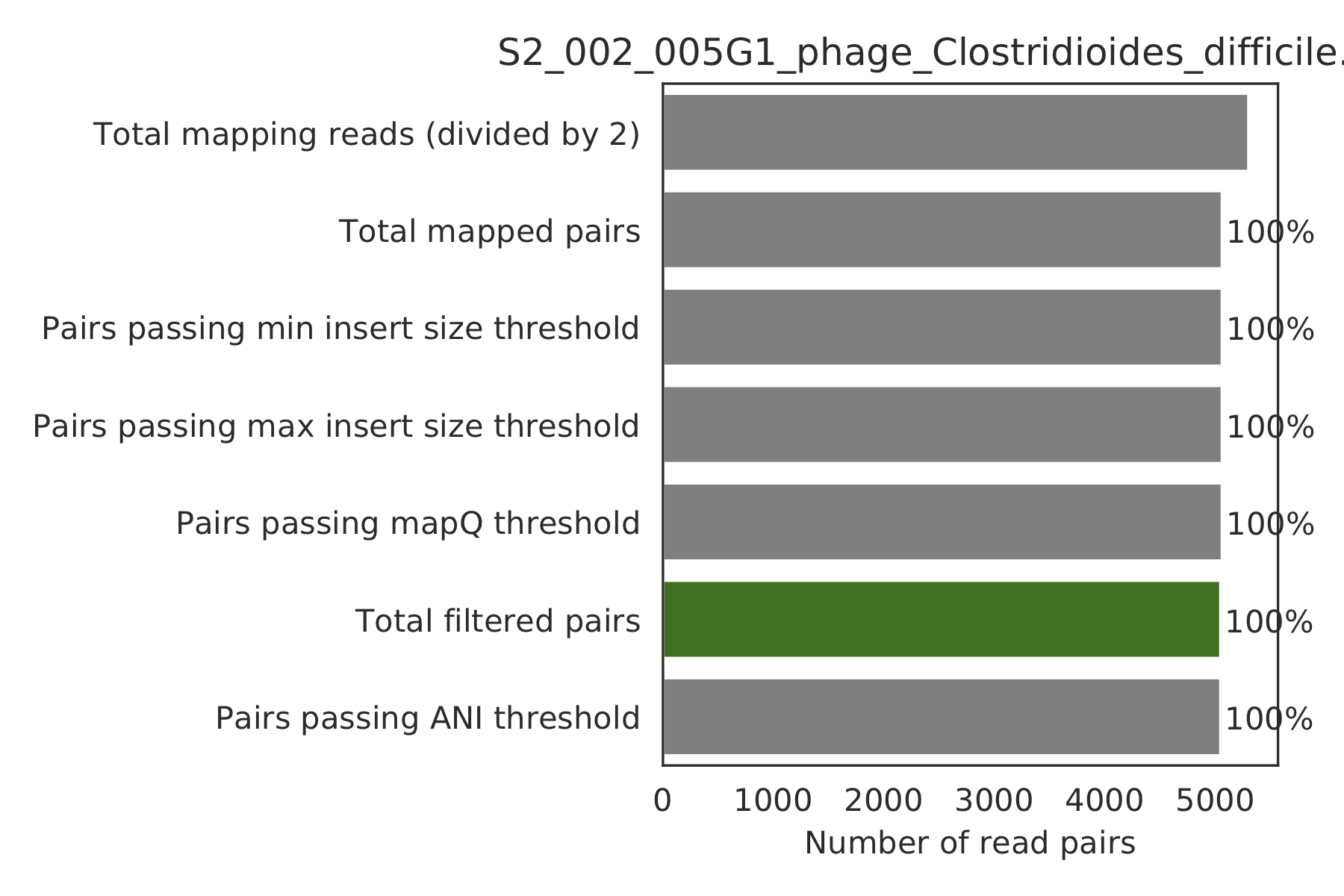
Bar plots showing how many reads got filtered out during filtering. All percentages are based on the number of paired reads; for an idea of how many reads were filtered out for being non-paired, compare the top bar and the second to top bar.
7) Scaffold inspection plot (large)
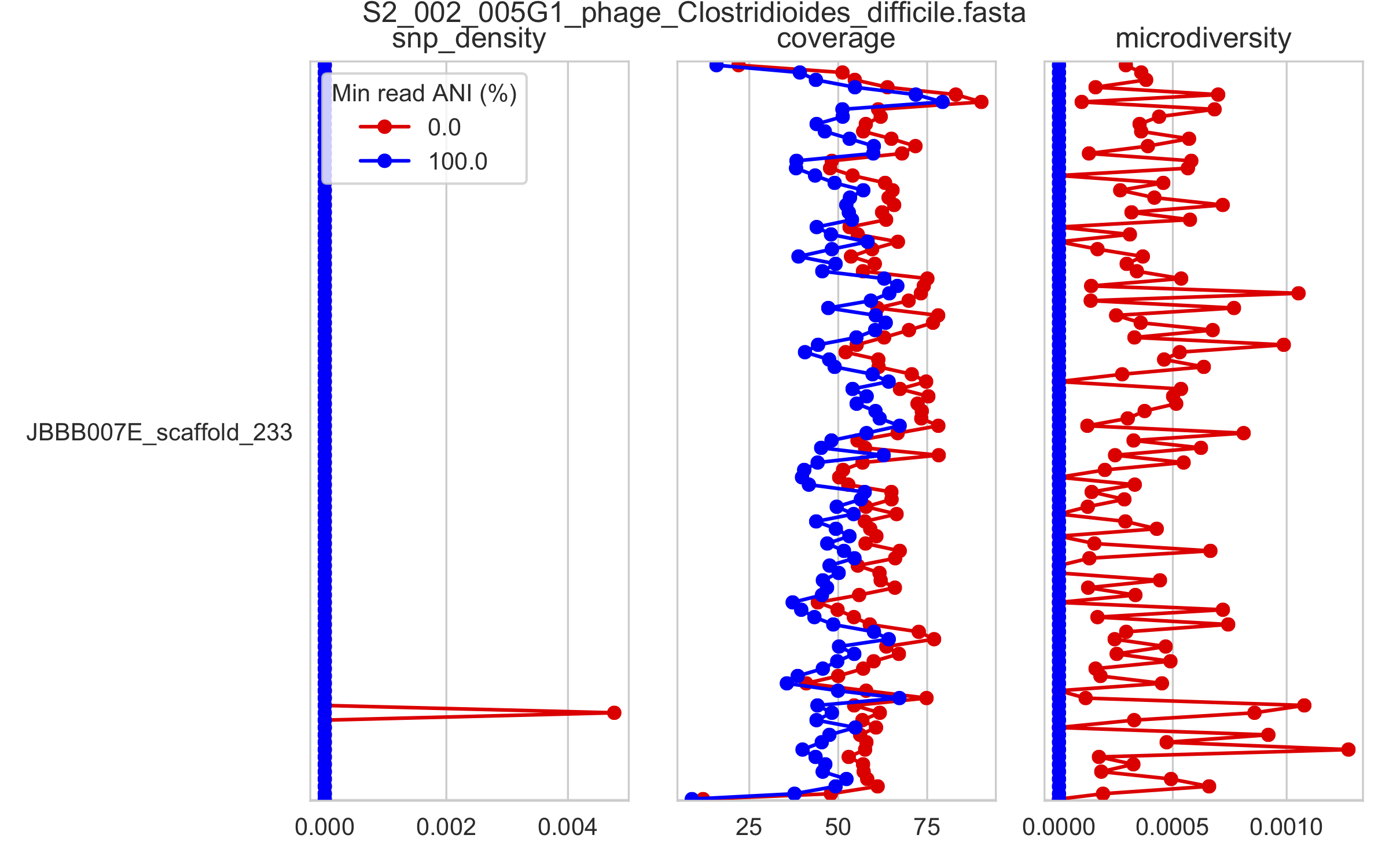
This is an elongated version of the genome-wide microdiversity metrics that is long enough for you to read scaffold names on the y-axis
8) Linkage with SNP type (GENES REQUIRED)
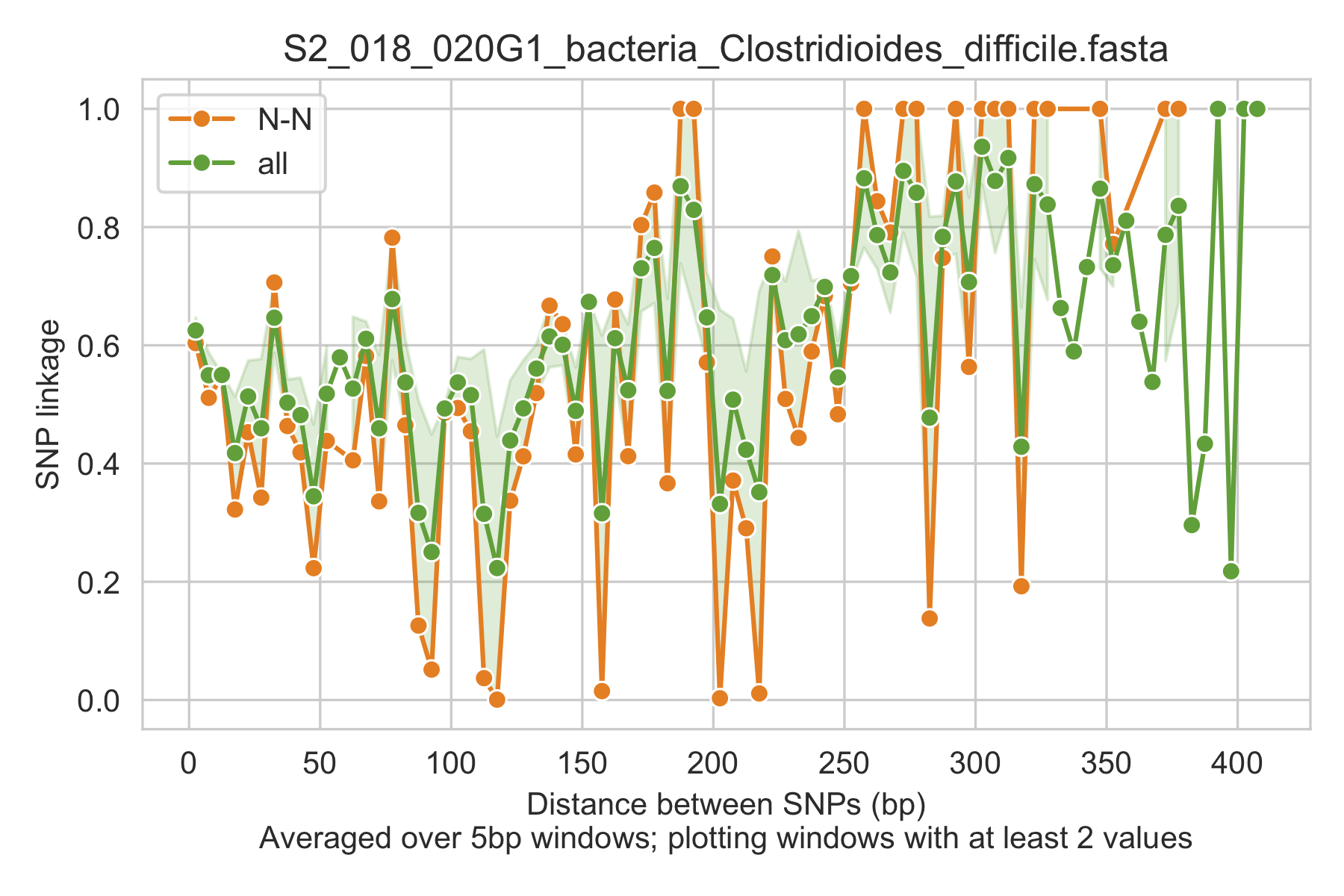
Linkage plot for pairs of non-synonymous SNPs and all pairs of SNPs
9) Gene histograms (GENES REQUIRED)
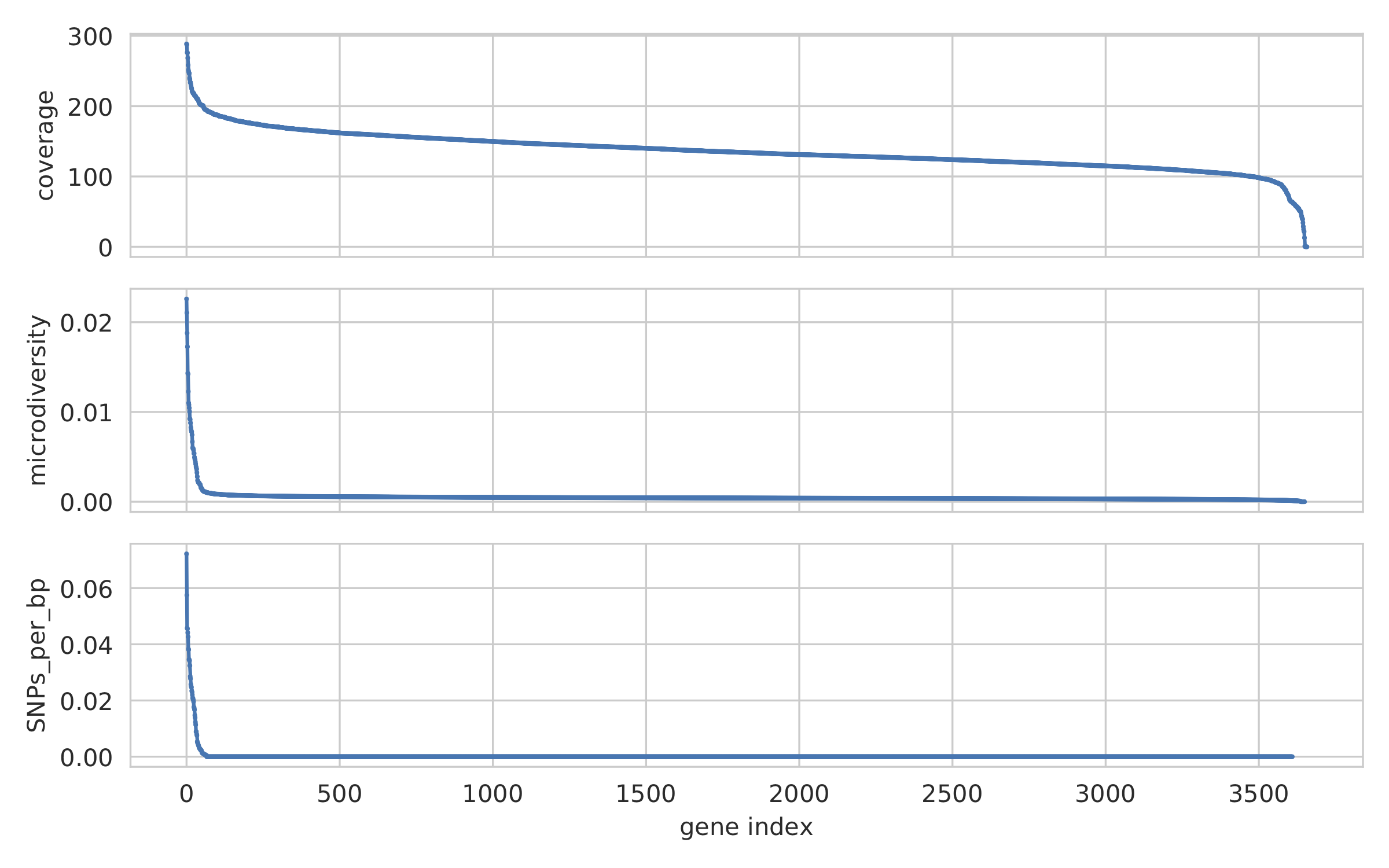
Histogram of values for all genes profiled
10) Compare dendrograms (RUN ON COMPARE; NOT PROFILE)
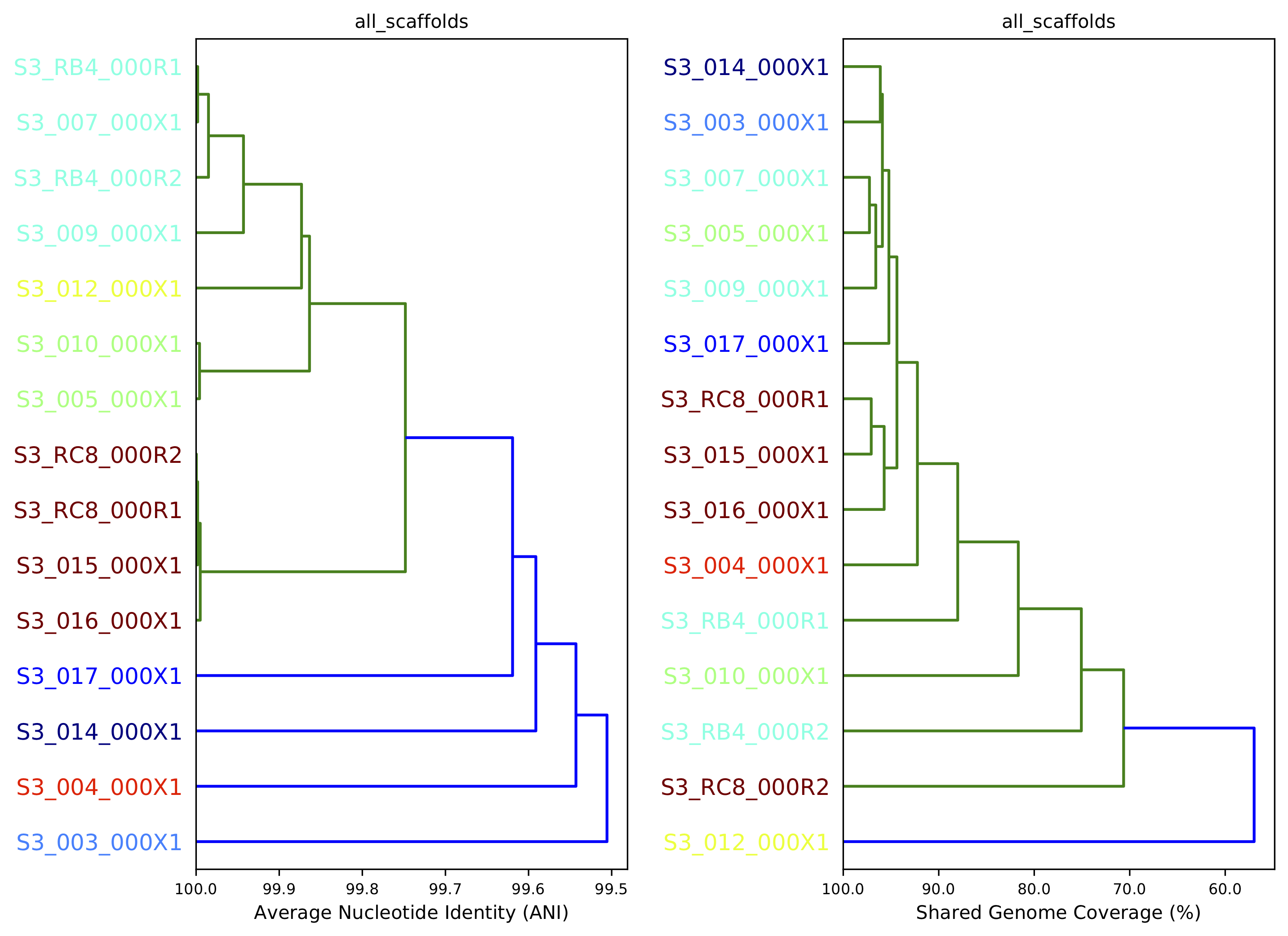
A dendrogram comparing all samples based on popANI and based on shared_bases.