Example output and explanations¶
InStrain produces a variety of output in the IS folder depending on which operations are run. Generally, output that is meant for human eyes to be easily interpretable is located in the output folder.
inStrain profile¶
A typical run of inStrain will yield the following files in the output folder:
scaffold_info.tsv¶
This gives basic information about the scaffolds in your sample at the highest allowed level of read identity.
| scaffold | length | breadth | coverage | coverage_median | coverage_std | bases_w_0_coverage | mean_clonality | median_clonality | mean_microdiversity | median_microdiversity | unmaskedBreadth | breadth_expected | SNPs | Referece_SNPs | BiAllelic_SNPs | MultiAllelic_SNPs | consensus_SNPs | population_SNPs | conANI | popANI |
| S3_003_000X1_scaffold_21039 | 1049 | 0.9609151572926596 | 7.778836987607247 | 9 | 3.6242339424115295 | 41 | 0.9984115827692688 | 1.0 | 0.0015884172307313313 | 0.0 | 0.7836034318398475 | 0.9989601856174312 | 1 | 0 | 1 | 0 | 0 | 0 | 1.0 | 1.0 |
| S3_003_000X1_scaffold_21063 | 1048 | 0.3721374045801527 | 0.6698473282442748 | 0 | 0.978669048484894 | 658 | 0.0 | 0.4464898509344126 | 0 | 0 | 0 | 0 | 0 | 0 | 0.0 | 0.0 | ||||
| S3_003_000X1_scaffold_21081 | 1047 | 0.2082139446036294 | 0.2082139446036294 | 0 | 0.4060306612513717 | 829 | 0.0 | 0.1679418203027453 | 0 | 0 | 0 | 0 | 0 | 0 | 0.0 | 0.0 | ||||
| S3_003_000X1_scaffold_21188 | 1043 | 0.3547459252157239 | 0.4688398849472674 | 0 | 0.6908089219842111 | 673 | 0.0 | 0.338989542420026 | 0 | 0 | 0 | 0 | 0 | 0 | 0.0 | 0.0 | ||||
| S3_003_000X1_scaffold_21225 | 1042 | 0.7821497120921305 | 4.341650671785029 | 5 | 3.4491608427332947 | 227 | 1.0 | 1.0 | 0.0 | 0.0 | 0.5374280230326296 | 0.9783700757950428 | 0 | 0 | 0 | 0 | 0 | 0 | 1.0 | 1.0 |
- scaffold
- The name of the scaffold in the input .fasta file
- length
- Full length of the scaffold in the input .fasta file
- breadth
- The percentage of bases in the scaffold that are covered by at least a single read. A breadth of 1 means that all bases in the scaffold have at least one read covering them
- coverage
- The average depth of coverage on the scaffold. If half the bases in a scaffold have 5 reads on them, and the other half have 10 reads, the coverage of the scaffold will be 7.5
- coverage_median
- The median coverage value of all bases in the scaffold, included bases with 0 coverage
- coverage_std
- The standard deviation of all coverage values
- bases_w_0_coverage
- The number of bases with 0 coverage
- mean_clonality
- The mean clonality value of all bases in the scaffold that have a clonality value calculated. So if only 1 base on the scaffold meats the minimum coverage to calculate clonality, the mean_clonality of the scaffold will be the clonality of that base
- median_clonality
- The median clonality value of all bases in the scaffold that have a clonality value calculated
- mean_microdiversity
- The mean mean_microdiversity value of all bases in the scaffold that have a mean_microdiversity value calculated (microdiveristy = 1 - clonality)
- median_microdiversity
- The median microdiversity value of all bases in the scaffold that have a microdiversity value calculated
- unmaskedBreadth
- The percentage of bases in the scaffold that have at least the min_cov number of bases. This value multiplied by the length of the scaffold gives the percentage of bases for which clonality is calculated and on which SNPs can be called
- SNPs
- The total number of SNPs called on this scaffold
- breadth_expected
- This tells you the breadth that you should expect if reads are evenly distributed along the genome, given the reported coverage value. Based on the function breadth = -1.000 * e^(0.883 * coverage) + 1.000. This is useful to establish whether or not the scaffold is actually in the reads, or just a fraction of the scaffold. If your coverage is 10x, the expected breadth will be ~1. If your actual breadth is significantly lower then the expected breadth, this means that reads are mapping only to a specific region of your scaffold (transposon, etc.)
- SNPs
- The total number of SNPs called on this scaffold
- Referece_SNPs
- The number of SNPs called on this scaffold with allele_count = 1. This means that the only allele detected in the reads is different from the reference base
- BiAllelic_SNPs
- The number of SNPs called on this scaffold with allele_count = 2. This means that there are two possible alleles at this position
- MultiAllelic_SNPs
- The number of SNPs called on this scaffold with allele_count > 2. This means that there are more than two possible alleles at this position
- consensus_SNPs
- The number of SNPs called on this scaffold with allele_count > 0 and where consensus base is not the reference base. This should be the same as Reference_SNPs under almost all circumstances
- population_SNPs
- These are SNPs where the reference base isn’t detected at all, regardless of the allele count.
- conANI
- The average nucleotide identity between the reads in the sample and the .fasta file based on consensus SNPs. Calculated using the formula ANI = (unmaskedBreadth * length) - consensus_SNPs)/ (unmaskedBreadth * length))
- popANI
- The average nucleotide identity between the reads in the sample and the .fasta file based on consensus SNPs. Calculated using the formula ANI = (unmaskedBreadth * length) - population_SNPs)/ (unmaskedBreadth * length))
mapping_info.tsv¶
This provides an overview of the number of reads that map to each scaffold, and some basic metrics about their quality.
| scaffold | unfiltered_reads | unfiltered_pairs | pass_filter_cutoff | pass_max_insert | pass_min_insert | pass_min_mapq | filtered_pairs | mean_mistmaches | mean_insert_distance | mean_mapq_score | mean_pair_length | median_insert | mean_PID |
| all_scaffolds | 3802370 | 1790817 | 1674511 | 1784011 | 1790699 | 1790817 | 1668496 | 2.7480758782164787 | 293.0713925543481 | 23.46918082640493 | 298.38404705785126 | 246.0 | 0.9906729188638016 |
| S3_002_000X1_scaffold_1162 | 12 | 6 | 6 | 6 | 6 | 6 | 6 | 1.0 | 281.1666666666667 | 25.16666666666667 | 300.0 | 287.0 | 0.9966666666666668 |
| S3_002_000X1_scaffold_1005 | 10 | 5 | 5 | 5 | 5 | 5 | 5 | 0.2 | 318.0 | 33.2 | 299.8 | 208.0 | 0.9993333333333332 |
| S3_002_000X1_scaffold_1151 | 6 | 3 | 3 | 3 | 3 | 3 | 3 | 5.666666666666668 | 280.3333333333333 | 19.666666666666668 | 300.0 | 293.0 | 0.9811111111111112 |
| S3_002_000X1_scaffold_1004 | 14 | 6 | 6 | 6 | 6 | 6 | 6 | 0.5 | 295.5 | 16.666666666666668 | 300.0 | 248.0 | 0.9983333333333334 |
The following metrics are provided for all individual scaffolds, and for all scaffolds together (scaffold “all_scaffolds”). For the max insert cutoff, the median_insert for all_scaffolds is used
- header line
- The header line (starting with #; not shown in the above table) describes the parameters that were used to filter the reads
- scaffold
- The name of the scaffold in the input .fasta file
- unfiltered_reads
- The raw number of reads that map to this scaffold
- unfiltered_pairs
- The raw number of pairs of reads that map to this scaffold. Only paired reads are used by inStrain
- pass_filter_cutoff
- The number of pairs of reads mapping to this scaffold that pass the ANI filter cutoff (specified in the header as “filter_cutoff”)
- pass_max_insert
- The number of pairs of reads mapping to this scaffold that pass the maximum insert size cutoff- that is, their insert size is less than 3x the median insert size of all_scaffolds. Note that the insert size is measured from the start of the first read to the end of the second read (2 perfectly overlapping 50bp reads will have an insert size of 50bp)
- pass_min_insert
- The number of pairs of reads mapping to this scaffold that pass the minimum insert size cutoff
- pass_min_mapq
- The number of pairs of reads mapping to this scaffold that pass the minimum mapQ score cutoff
- filtered_pairs
- The number of pairs of reads that pass all cutoffs
- mean_mistmaches
- Among all pairs of reads mapping to this scaffold, the mean number of mismatches
- mean_insert_distance
- Among all pairs of reads mapping to this scaffold, the mean insert distance. Note that the insert size is measured from the start of the first read to the end of the second read (2 perfectly overlapping 50bp reads will have an insert size of 50bp)
- mean_mapq_score
- Among all pairs of reads mapping to this scaffold, the average mapQ score
- mean_pair_length
- Among all pairs of reads mapping to this scaffold, the average length of both reads in the pair summed together
- median_insert
- Among all pairs of reads mapping to this scaffold, the median insert distance.
- mean_PID
- Among all pairs of reads mapping to this scaffold, the average percentage ID of both reads in the pair to the reference .fasta file
SNVs.tsv¶
This describes the SNPs that are detected in this mapping.
| scaffold | position | ref_base | A | C | T | G | con_base | var_base | allele_count | cryptic | position_coverage | var_freq | ref_freq |
| S3_003_000X1_scaffold_21039 | 833 | C | 2 | 7 | 0 | 0 | C | A | 2 | False | 9 | 0.2222222222222222 | 0.7777777777777778 |
| S3_003_000X1_scaffold_20 | 99 | C | 0 | 0 | 5 | 0 | T | A | 1 | False | 5 | 0.0 | 1.0 |
| S3_003_000X1_scaffold_20 | 123 | A | 0 | 0 | 0 | 11 | G | A | 1 | False | 11 | 0.0 | 1.0 |
| S3_003_000X1_scaffold_20 | 261 | T | 19 | 0 | 0 | 0 | A | A | 1 | False | 19 | 1.0 | 1.0 |
| S3_003_000X1_scaffold_20 | 291 | C | 0 | 16 | 2 | 0 | C | T | 2 | False | 18 | 0.1111111111111111 | 0.8888888888888888 |
See the module_descriptions for what constitutes a SNP (what makes it into this table)
- scaffold
- The scaffold that the SNP is on
- position
- The genomic position of the SNP
- ref_base
- The reference base in the .fasta file at that position
- A, C, T, and G
- The number of mapped reads encoding each of the bases
- con_base
- The consensus base; the base that is supported by the most reads
- var_base
- Variant base; the base with the second most reads
- morphia
- The number of bases that are detected above background levels. In order to be detected above background levels, you must pass an fdr filter. See module descriptions for a description of how that works. A morphia of 0 means no bases are supported by the reads, a morphia of 1 means that only 1 base is supported by the reads, a morphia of 2 means two bases are supported by the reads, etc.
- cryptic
- If a SNP is cryptic, it means that it is detected when using a lower read mismatch threshold, but becomes undetected when you move to a higher read mismatch level. See “dealing with mm” in the advanced_use section for more details on what this means.
- position_coverage
- The total number of reads at this position
- var_freq
- The fraction of reads supporting the var_base
- ref_freq
- The fraction of reds supporting the ref_base
- con_freq
- The fraction of reds supporting the con_base
linkage.tsv¶
This describes the linkage between pairs of SNPs in the mapping that are found on the same read pair at least min_snp times.
| r2 | d_prime | r2_normalized | d_prime_normalized | total | countAB | countAb | countaB | countab | allele_A | allele_a | allele_B | allele_b | distance | position_A | position_B | scaffold |
| 1.0 | 1.0 | 1.0 | 1.0 | 27 | 0 | 14 | 13 | 0 | G | A | T | C | 45 | 191425 | 191470 | S3_003_000X1_scaffold_20 |
| 0.10743801652892566 | 1.0000000000000002 | 0.05263157894736843 | 1.0 | 24 | 13 | 0 | 9 | 2 | G | A | C | A | 80 | 191425 | 191505 | S3_003_000X1_scaffold_20 |
| 0.08333333333333348 | 1.0 | 0.07894736842105264 | 1.0 | 26 | 11 | 2 | 13 | 0 | T | C | C | A | 35 | 191470 | 191505 | S3_003_000X1_scaffold_20 |
| 1.0000000000000009 | 1.0 | 1.0 | 1.0 | 30 | 22 | 0 | 0 | 8 | C | T | T | C | 12 | 99342 | 99354 | S3_003_000X1_scaffold_88 |
| 1.0000000000000004 | 1.0 | 1.0 | 1.0 | 22 | 17 | 0 | 0 | 5 | C | T | T | A | 60 | 99342 | 99402 | S3_003_000X1_scaffold_88 |
Linkage is used primarily to determine if organisms are undergoing horizontal gene transfer or not. It’s calculated for pairs of SNPs that can be connected by at least min_snp reads. It’s based on the assumption that each SNP as two alleles (for example, a A and b B). This all gets a bit confusing and has a large amount of literature around each of these terms, but I’ll do my best to briefly explain what’s going on
- scaffold
- The scaffold that both SNPs are on
- position_A
- The position of the first SNP on this scaffold
- position_B
- The position of the second SNP on this scaffold
- distance
- The distance between the two SNPs
- allele_A
- One of the two bases at position_A
- allele_a
- The other of the two bases at position_A
- allele_B
- One of the bases at position_B
- allele_b
- The other of the two bases at position_B
- countAB
- The number of read-pairs that have allele_A and allele_B
- countAb
- The number of read-pairs that have allele_A and allele_b
- countaB
- The number of read-pairs that have allele_a and allele_B
- countab
- The number of read-pairs that have allele_a and allele_b
- total
- The total number of read-pairs that have have information for both position_A and position_B
- r2
- This is the r-squared linkage metric. See below for how it’s calculated
- d_prime
- This is the d-prime linkage metric. See below for how it’s calculated
- r2_normalized, d_prime_normalized
- These are calculated by rarefying to
min_snpnumber of read pairs. See below for how it’s calculated
Python code for the calculation of these metrics:
freq_AB = float(countAB) / total
freq_Ab = float(countAb) / total
freq_aB = float(countaB) / total
freq_ab = float(countab) / total
freq_A = freq_AB + freq_Ab
freq_a = freq_ab + freq_aB
freq_B = freq_AB + freq_aB
freq_b = freq_ab + freq_Ab
linkD = freq_AB - freq_A * freq_B
if freq_a == 0 or freq_A == 0 or freq_B == 0 or freq_b == 0:
r2 = np.nan
else:
r2 = linkD*linkD / (freq_A * freq_a * freq_B * freq_b)
linkd = freq_ab - freq_a * freq_b
# calc D-prime
d_prime = np.nan
if (linkd < 0):
denom = max([(-freq_A*freq_B),(-freq_a*freq_b)])
d_prime = linkd / denom
elif (linkD > 0):
denom = min([(freq_A*freq_b), (freq_a*freq_B)])
d_prime = linkd / denom
################
# calc rarefied
rareify = np.random.choice(['AB','Ab','aB','ab'], replace=True, p=[freq_AB,freq_Ab,freq_aB,freq_ab], size=min_snp)
freq_AB = float(collections.Counter(rareify)['AB']) / min_snp
freq_Ab = float(collections.Counter(rareify)['Ab']) / min_snp
freq_aB = float(collections.Counter(rareify)['aB']) / min_snp
freq_ab = float(collections.Counter(rareify)['ab']) / min_snp
freq_A = freq_AB + freq_Ab
freq_a = freq_ab + freq_aB
freq_B = freq_AB + freq_aB
freq_b = freq_ab + freq_Ab
linkd_norm = freq_ab - freq_a * freq_b
if freq_a == 0 or freq_A == 0 or freq_B == 0 or freq_b == 0:
r2_normalized = np.nan
else:
r2_normalized = linkd_norm*linkd_norm / (freq_A * freq_a * freq_B * freq_b)
# calc D-prime
d_prime_normalized = np.nan
if (linkd_norm < 0):
denom = max([(-freq_A*freq_B),(-freq_a*freq_b)])
d_prime_normalized = linkd_norm / denom
elif (linkd_norm > 0):
denom = min([(freq_A*freq_b), (freq_a*freq_B)])
d_prime_normalized = linkd_norm / denom
rt_dict = {}
for att in ['r2', 'd_prime', 'r2_normalized', 'd_prime_normalized', 'total', 'countAB', \
'countAb', 'countaB', 'countab', 'allele_A', 'allele_a', \
'allele_B', 'allele_b']:
rt_dict[att] = eval(att)
inStrain compare¶
A typical run of inStrain will yield the following files in the output folder:
| scaffold | name1 | name2 | coverage_overlap | compared_bases_count | percent_genome_compared | length | consensus_SNPs | population_SNPs | conANI | popANI |
| S3_016_000X1_scaffold_14208 | Sloan3AllGenomeInventory.fasta-vs-S3_003_000X1.sorted.bam | Sloan3AllGenomeInventory.fasta-vs-S3_016_000X1.sorted.bam | 0.9825304393859184 | 1856 | 0.9814912744579588 | 1891 | 7 | 0 | 0.996228448275862 | 1.0 |
| S3_016_000X1_scaffold_9493 | Sloan3AllGenomeInventory.fasta-vs-S3_003_000X1.sorted.bam | Sloan3AllGenomeInventory.fasta-vs-S3_016_000X1.sorted.bam | 0.9778541428025964 | 2561 | 0.977107974055704 | 2621 | 2 | 0 | 0.9992190550566185 | 1.0 |
| S3_016_000X1_scaffold_12686 | Sloan3AllGenomeInventory.fasta-vs-S3_003_000X1.sorted.bam | Sloan3AllGenomeInventory.fasta-vs-S3_016_000X1.sorted.bam | 0.9787336877718704 | 2025 | 0.9768451519536904 | 2073 | 7 | 0 | 0.9965432098765432 | 1.0 |
| S3_016_000X1_scaffold_11829 | Sloan3AllGenomeInventory.fasta-vs-S3_003_000X1.sorted.bam | Sloan3AllGenomeInventory.fasta-vs-S3_016_000X1.sorted.bam | 0.9739130434782608 | 2128 | 0.9712460063897764 | 2191 | 14 | 0 | 0.9934210526315792 | 1.0 |
| S3_016_000X1_scaffold_8891 | Sloan3AllGenomeInventory.fasta-vs-S3_003_000X1.sorted.bam | Sloan3AllGenomeInventory.fasta-vs-S3_016_000X1.sorted.bam | 0.9826212889210716 | 2714 | 0.9826212889210716 | 2762 | 5 | 0 | 0.9981577008106116 | 1.0 |
- scaffold
- The scaffold being compared
- name1
- The name of the first inStrain profile being compared
- name2
- The name of the second inStrain profile being compared
- coverage_overlap
- The percentage of bases that are either covered or not covered in both of the profiles (covered = the base is present at at least min_snp coverage). The formula is length(coveredInBoth) / length(coveredInEither). If both scaffolds have 0 coverage, this will be 0.
- compared_bases_count
- The number of considered bases; that is, the number of bases with at least min_snp coverage in both profiles. Formula is length([x for x in overlap if x == True]).
- percent_genome_compared
- The percentage of bases in the scaffolds that are covered by both. The formula is length([x for x in overlap if x == True])/length(overlap). When ANI is np.nan, this must be 0. If both scaffolds have 0 coverage, this will be 0.
- length
- The total length of the scaffold
- consensus_SNPs
- The number of locations along the genome where both samples have the base at >= 5x coverage, and the consensus allele in each sample is different
- population_SNPs
- The number of locations along the genome where both samples have the base at >= 5x coverage, and no alleles are shared between either sample. See inStrain manuscript for more details.
- popANI
- The average nucleotide identity among compared bases between the two scaffolds, based on population_SNPs. Calculated using the formula popANI = (compared_bases_count - population_SNPs) / compared_bases_count
- conANI
- The average nucleotide identity among compared bases between the two scaffolds, based on consensus_SNPs. Calculated using the formula conANI = (compared_bases_count - consensus_SNPs) / compared_bases_count
inStrain profile_genes¶
A typical run of inStrain profile_genes will yield the following additional files in the output folder:
gene_info.tsv¶
This describes some basic information about the genes being profiled
| gene | scaffold | direction | partial | start | end | coverage | breadth | clonality | microdiversity | masked_breadth | SNPs_per_bp | min_ANI |
| S3_002_028G1_scaffold_0_1 | S3_002_028G1_scaffold_0 | -1 | False | 957 | 2219 | 0 | ||||||
| S3_002_028G1_scaffold_0_2 | S3_002_028G1_scaffold_0 | -1 | False | 2189 | 3136 | 0 | ||||||
| S3_002_028G1_scaffold_0_3 | S3_002_028G1_scaffold_0 | 1 | False | 3274 | 5013 | 0 | ||||||
| S3_002_028G1_scaffold_0_4 | S3_002_028G1_scaffold_0 | -1 | False | 5018 | 5746 | 0 | ||||||
| S3_002_028G1_scaffold_0_5 | S3_002_028G1_scaffold_0 | 1 | False | 5888 | 6862 | 0 |
- gene
- Name of the gene being profiled
- scaffold
- Scaffold that the gene is on
- direction
- Direction of the gene (based on prodigal call). If -1, means the gene is not coded in the direction expressed by the .fasta file
- partial
- If True this is a partial gene; based on not having partial=00 in the record description provided by Prodigal
- start
- Start of the gene (position on scaffold; 0-indexed)
- end
- End of the gene (position on scaffold; 0-indexed)
- coverage
- The mean coverage across the length of the gene
- breadth
- The number of bases in the gene that have at least 1x coverage
- microdiversity
- The mean nucleotide diversity (pi) among positions on the gene with at least 5x coverage
- clonality
- 1 - microdiversity
- masked_breadth
- The percentage of positions in the gene with at least 5x coverage
- SNPs_per_bp
- The number of positions on the gene where a SNP is called
- min_ANI
- The minimum read ANI level when profile_genes was run (0 means the value is whatever was set with Profile was originally run)
SNP_mutation_types.tsv¶
This describes whether SNPs are synonymous, nonsynonymous, or intergenic
| scaffold | position | ref_base | A | C | T | G | con_base | var_base | allele_count | position_coverage | var_freq | ref_freq | mutation_type | mutation | gene |
| S3_002_056W1_scaffold_121 | 2134 | C | 0 | 3 | 2 | 0 | C | T | 2 | 5 | 0.4 | 0.6 | N | N:H936Y | S3_002_056W1_scaffold_121_2 |
| S3_002_056W1_scaffold_121 | 8509 | G | 7 | 0 | 0 | 0 | A | A | 1 | 7 | 1.0 | 1.0 | N | N:G459R | S3_002_056W1_scaffold_121_11 |
| S3_002_056W1_scaffold_121 | 8510 | G | 7 | 0 | 0 | 0 | A | A | 1 | 7 | 1.0 | 1.0 | N | N:G460E | S3_002_056W1_scaffold_121_11 |
| S3_002_056W1_scaffold_121 | 16899 | G | 0 | 2 | 0 | 5 | G | C | 2 | 7 | 0.2857142857142857 | 0.7142857142857143 | N | N:G1068R | S3_002_056W1_scaffold_121_20 |
| S3_002_056W1_scaffold_121 | 24347 | C | 0 | 9 | 2 | 0 | C | T | 2 | 11 | 0.18181818181818185 | 0.8181818181818182 | N | N:Q894* | S3_002_056W1_scaffold_121_25 |
All genes with an allele_count of 1 or 2 make it into this table; see the above description of SNVs.tsv for details on what most of these columns mean
- mutation_type
- What type of mutation this is. N = nonsynonymous, S = synonymous, I = intergenic, M = there are multiple genes with this base so you cant tell
- mutation
- Short-hand code for the amino acid switch. If synonymous, this will be S: + the position. If nonsynonymous, this will be N: + the old amino acid + the position + the new amino acid.
- gene
- The gene this SNP is in
inStrain genome_wide¶
A typical run of inStrain genome_wide will yield the following additional files in the output folder:
genomeWide_scaffold_info.tsv¶
This is a genome-wide version of the scaffold report described above. See above for column descriptions.
| genome | detected_scaffolds | true_scaffolds | length | SNPs | Referece_SNPs | BiAllelic_SNPs | MultiAllelic_SNPs | consensus_SNPs | population_SNPs | breadth | coverage | coverage_std | mean_clonality | conANI | popANI | unmaskedBreadth | breadth_expected |
| S3_002_S3_002_000X1_S3_002_000X1_scaffold_633.fasta.fa | 1 | 1 | 19728 | 24 | 5 | 19 | 0 | 7 | 5 | 0.9462185725871858 | 4.5430859691808605 | 2.7106449701139903 | 0.998095248422326 | 0.9992792421746294 | 0.999485172981878 | 0.4922952149229522 | 0.9818945976123048 |
| S3_002_S3_002_000X1_S3_002_000X1_scaffold_980.fasta.fa | 1 | 1 | 11440 | 0 | 0 | 0 | 0 | 0 | 0 | 0.10113636363636364 | 0.10113636363636364 | 0.3015092031543595 | 0.0 | 0.0 | 0.0 | 0.08543195678460236 | |
| S3_002_S3_002_028Y1_S3_002_028Y1_scaffold_1.fasta.fa | 1 | 1 | 21455 | 0 | 0 | 0 | 0 | 0 | 0 | 0.5250058261477512 | 0.925378699603822 | 1.1239958370555831 | 0.9985388128180482 | 1.0 | 1.0 | 0.010207410859939408 | 0.5582933883068741 |
| S3_002_S3_002_028Y1_S3_002_028Y1_scaffold_22.fasta.fa | 1 | 1 | 15306 | 62 | 2 | 60 | 0 | 10 | 2 | 0.9562263164771984 | 4.977525153534561 | 4.1617488447219975 | 0.9939042740586184 | 0.9983668136534378 | 0.9996733627306876 | 0.4000392003136025 | 0.9876630284821302 |
| S3_002_S3_002_028Y1_S3_002_028Y1_scaffold_24.fasta.fa | 1 | 1 | 10383 | 64 | 6 | 58 | 0 | 18 | 6 | 0.9650390060676104 | 4.310507560435327 | 2.783478652159297 | 0.9912517160274896 | 0.9957865168539326 | 0.9985955056179776 | 0.4114417798324184 | 0.9777670126398924 |
genomeWide_mapping_info.tsv¶
This is a genome-wide version of the read report described above. See above for column descriptions.
| genome | scaffolds | unfiltered_reads | unfiltered_pairs | pass_filter_cutoff | pass_max_insert | pass_min_insert | pass_min_mapq | filtered_pairs | mean_mistmaches | mean_insert_distance | mean_mapq_score | mean_pair_length | median_insert | mean_PID |
| S2_002_005G1_phage_Clostridioides_difficile.fasta | 1 | 10605 | 5062 | 5048 | 5062 | 5062 | 5062 | 5048 | 0.3832477281706835 | 312.3638877913868 | 1.3024496246542872 | 293.6845120505729 | 308.0 | 0.998581261373412 |
| S2_018_020G1_bacteria_Clostridioides_difficile.fasta | 34 | 4453547 | 2163329 | 2149205 | 2163040 | 2162730 | 2163329 | 2148394 | 0.5636466689761853 | 321.3510672021471 | 41.47419579138972 | 293.33494491093336 | 312.5147058823529 | 0.9979527547934701 |
inStrain plot¶
This is what the results of inStrain plot look like.
1) Coverage and breadth vs. read mismatches¶
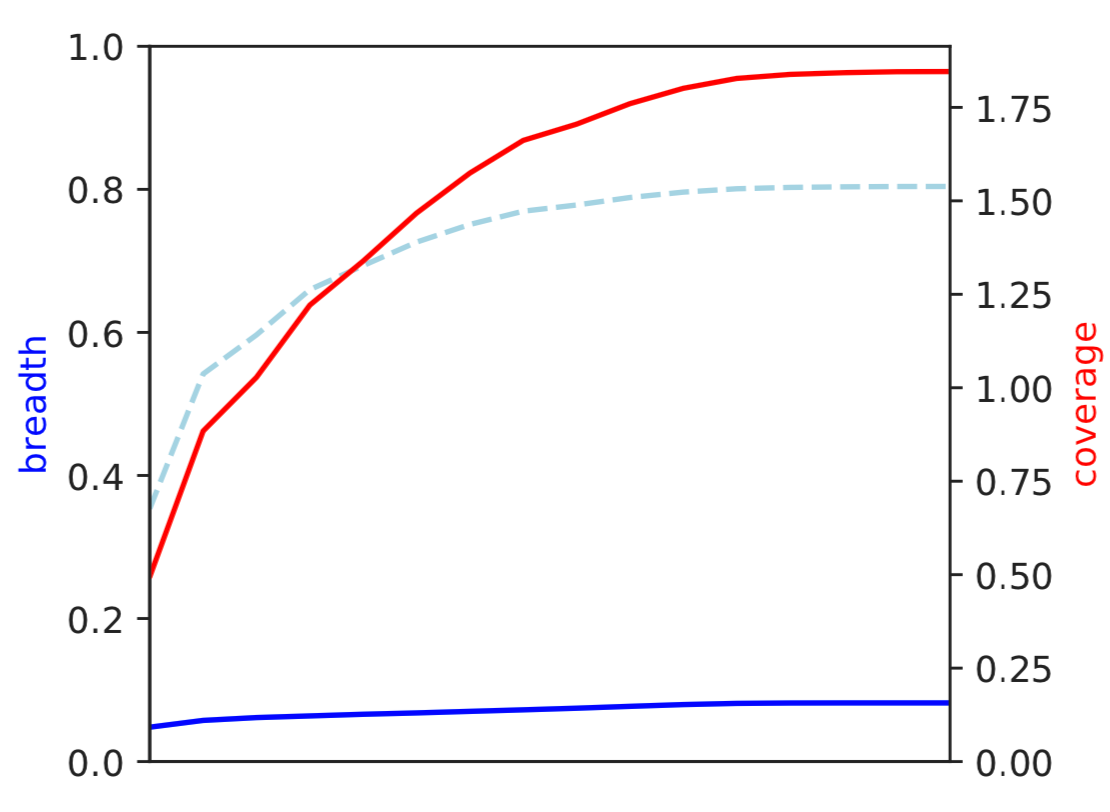
Breadth of coverage (blue line), coverage depth (red line), and expected breadth of coverage given the depth of coverage (dotted blue line) versus the minimum ANI of mapped reads. Coverage depth continues to increase while breadth of plateaus, suggesting that all regions of the reference genome are not present in the reads being mapped.
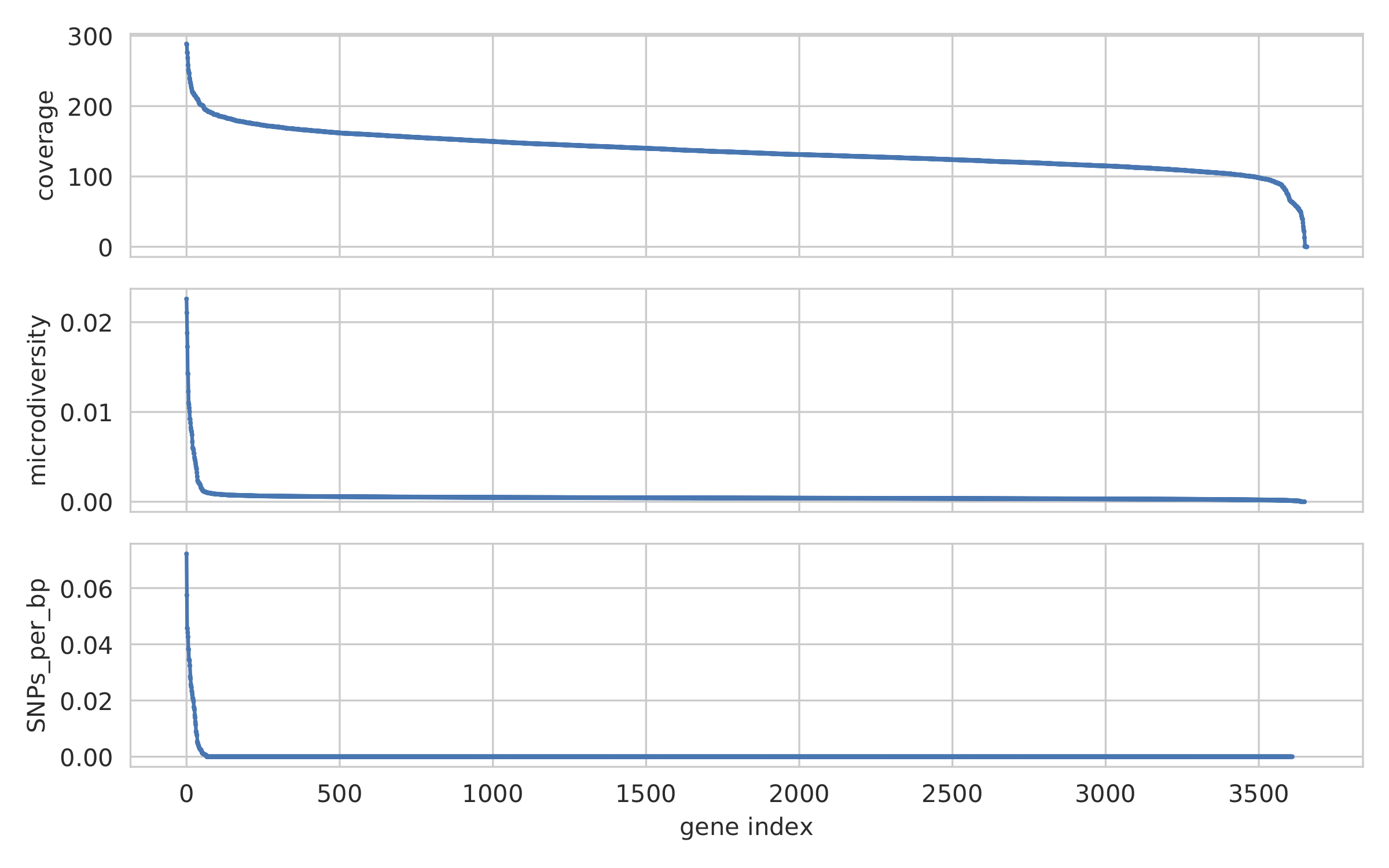
2) Genome-wide microdiversity metrics¶
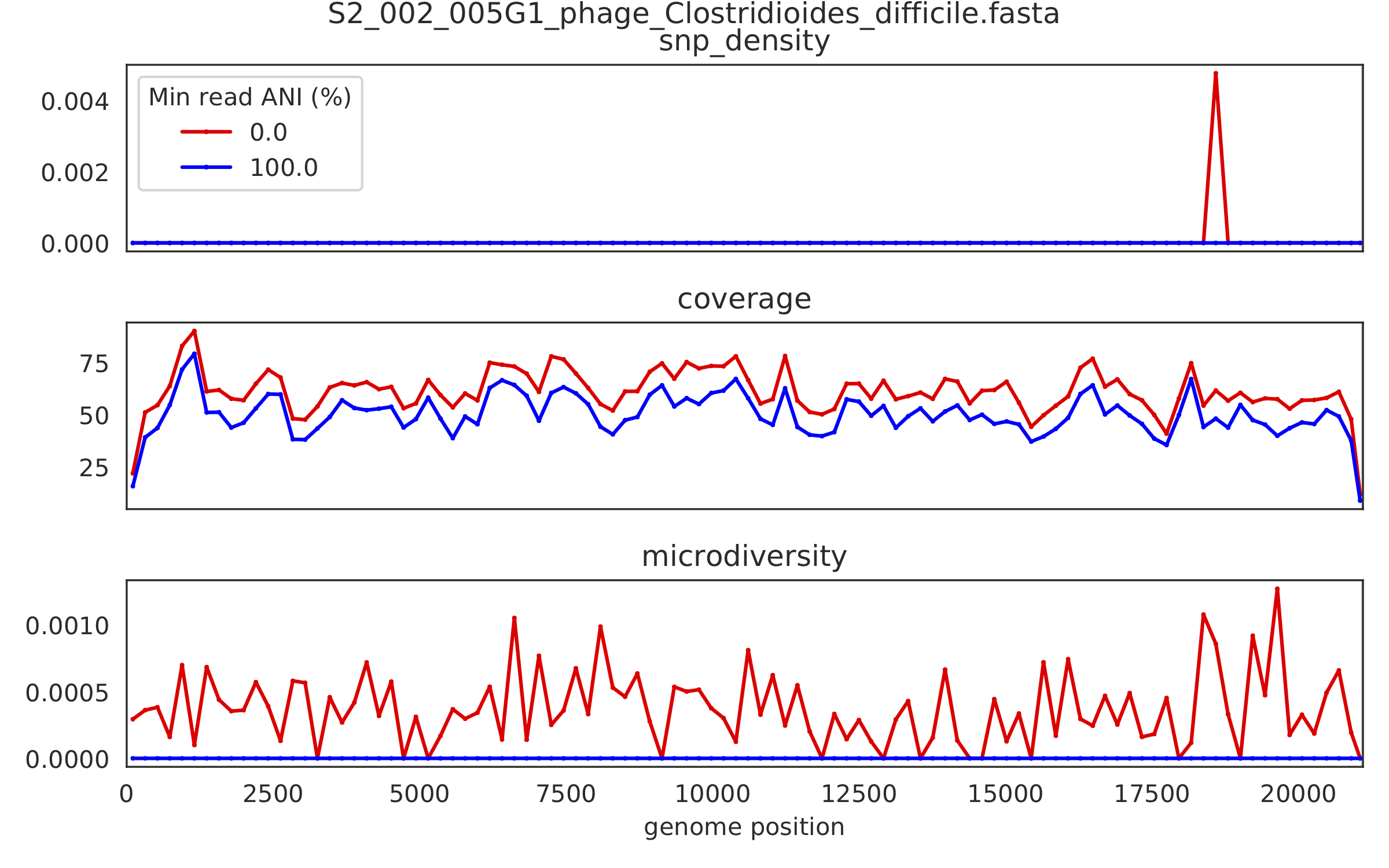
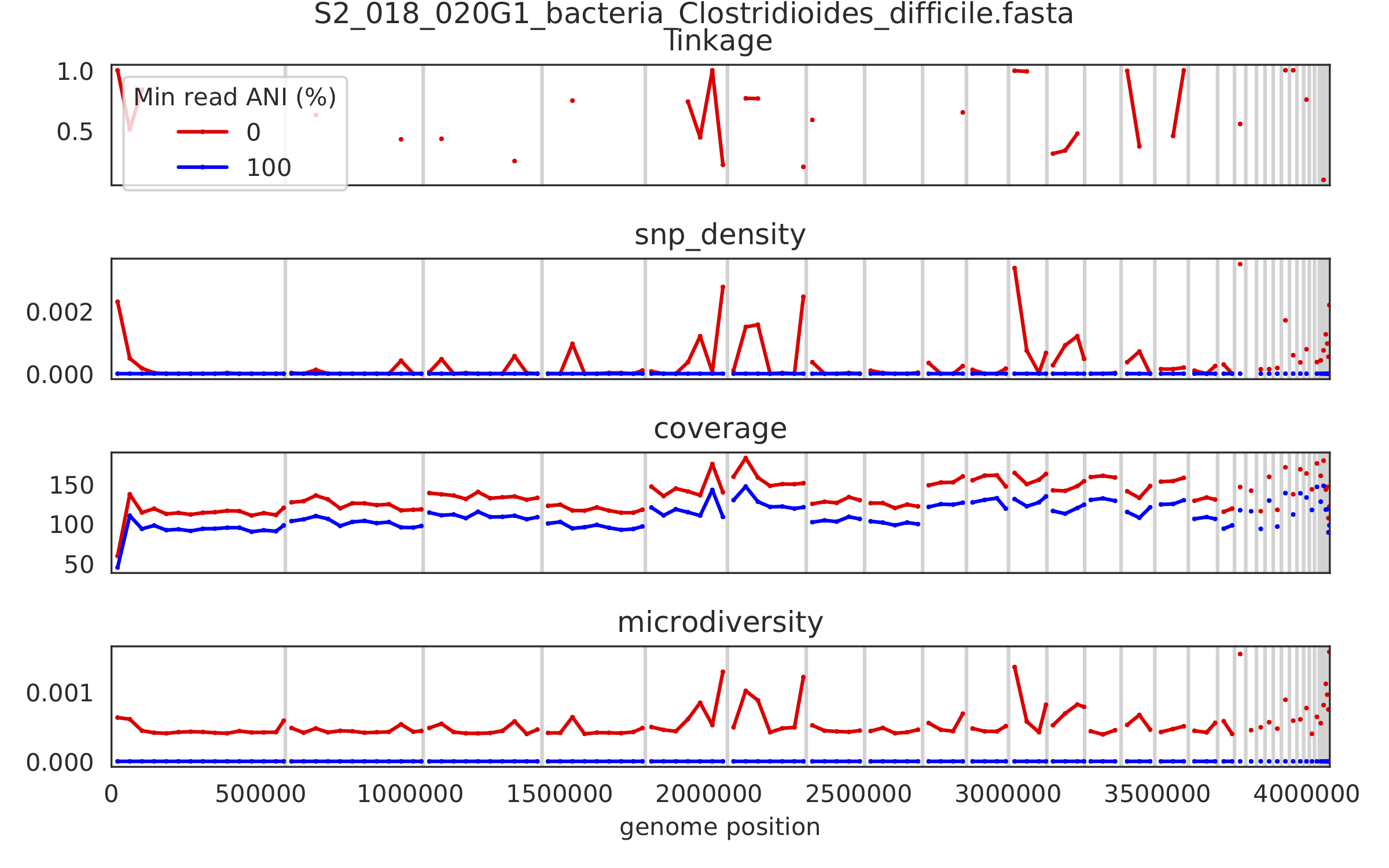
SNV density, coverage, and nucleotide diversity. Spikes in nucleotide diversity and SNV density do not correspond with increased coverage, indicating that the signals are not due to read mis-mapping. Positions with nucleotide diversity and no SNV-density are those where diversity exists but is not high enough to call a SNV
3) Read-level ANI distribution¶
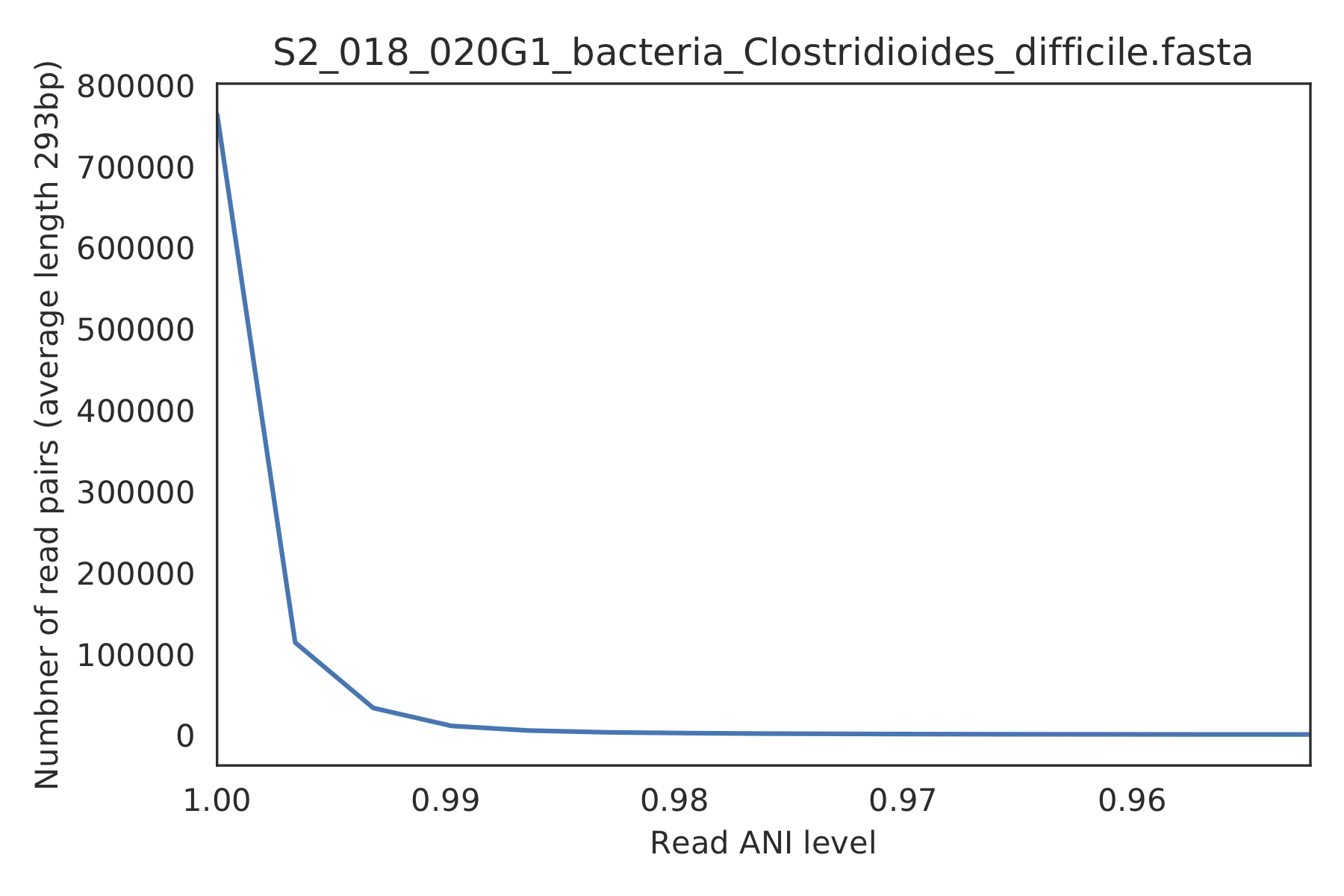
Distribution of read pair ANI levels when mapped to a reference genome; this plot suggests that the reference genome is >1% different than the mapped reads
4) Major allele frequencies¶
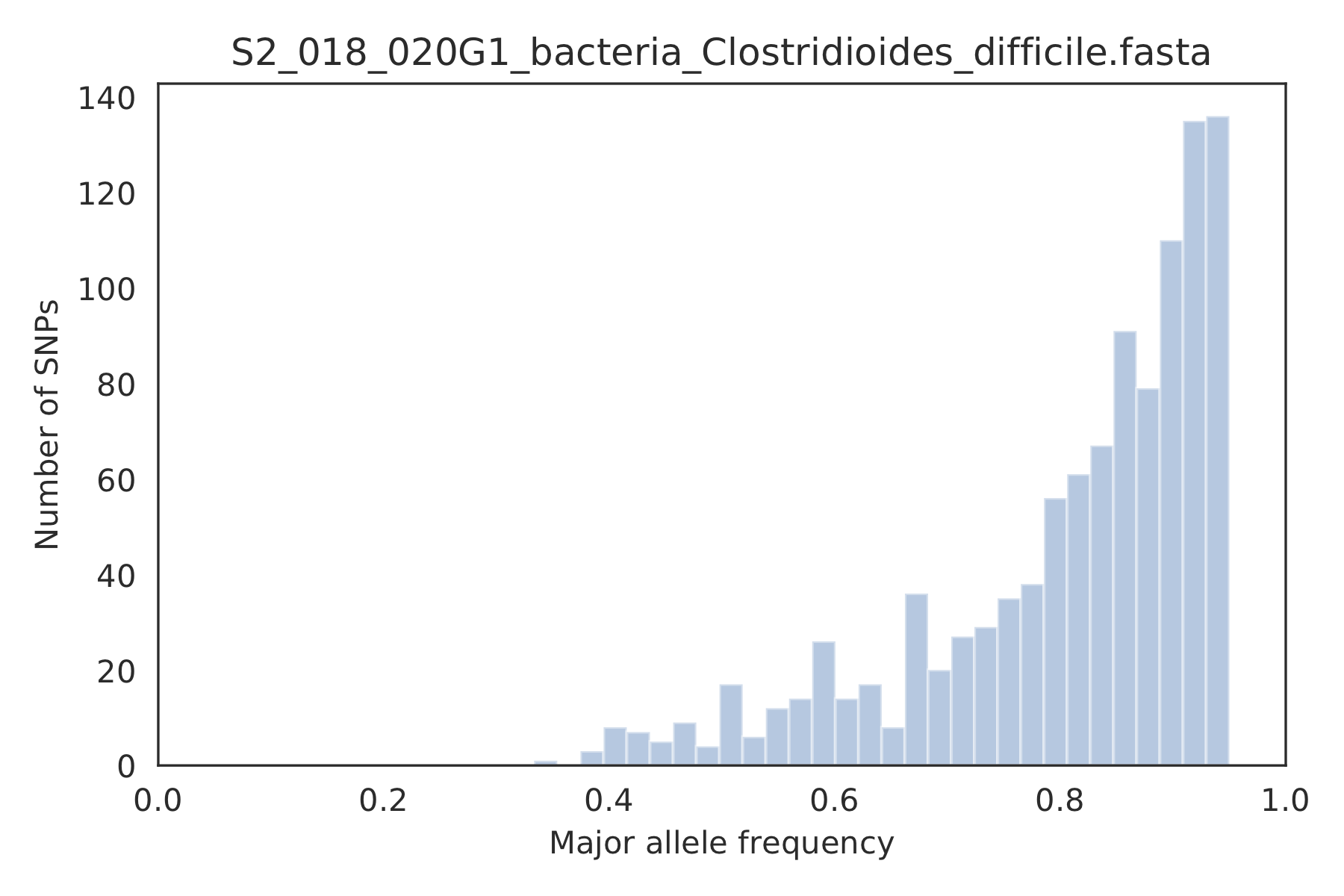
Distribution of the major allele frequencies of bi-allelic SNVs (the Site Frequency Spectrum). Alleles with major frequencies below 50% are the result of multiallelic sites. The lack of distinct puncta suggest that more than a few distinct strains are present.
5) Linkage decay¶
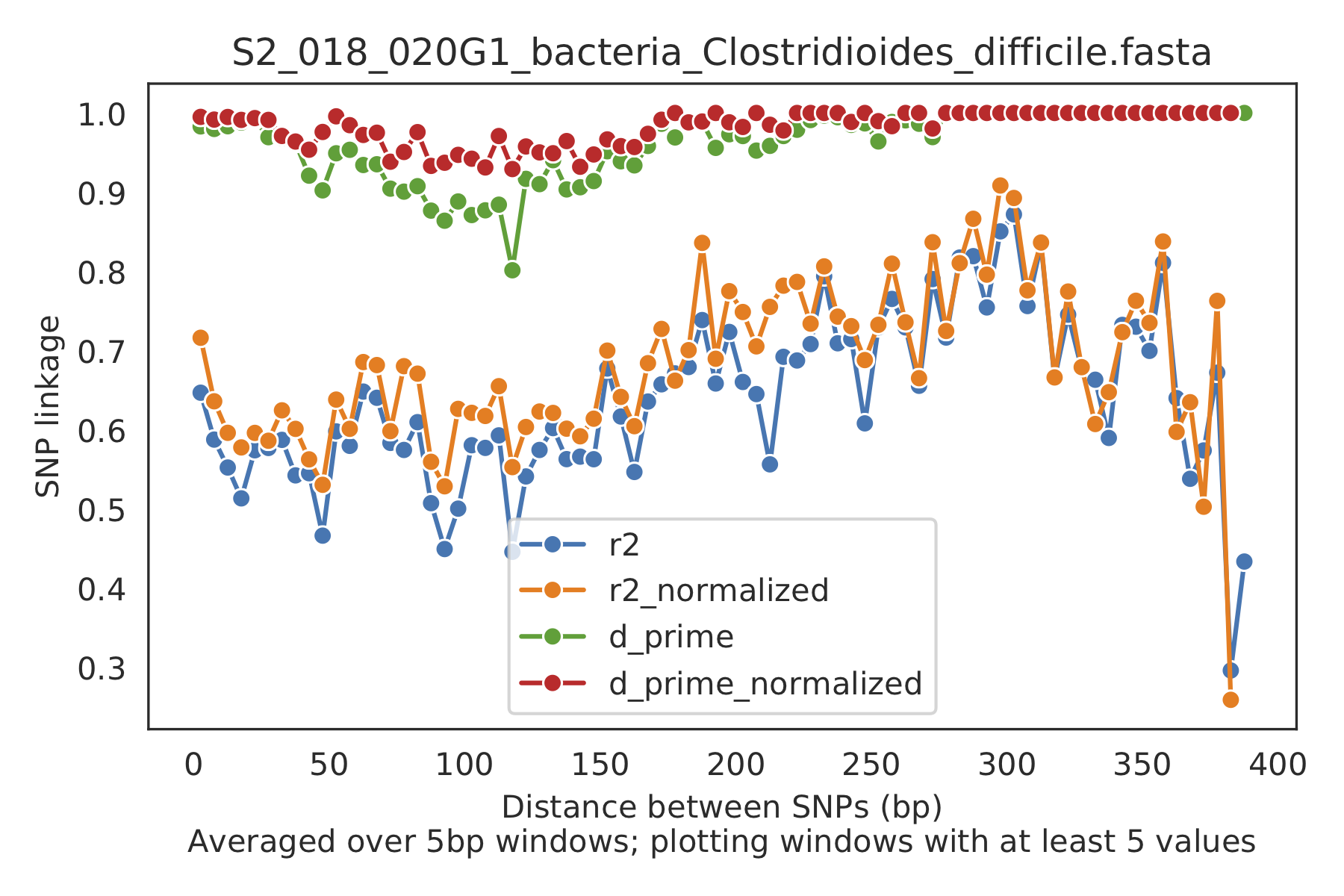
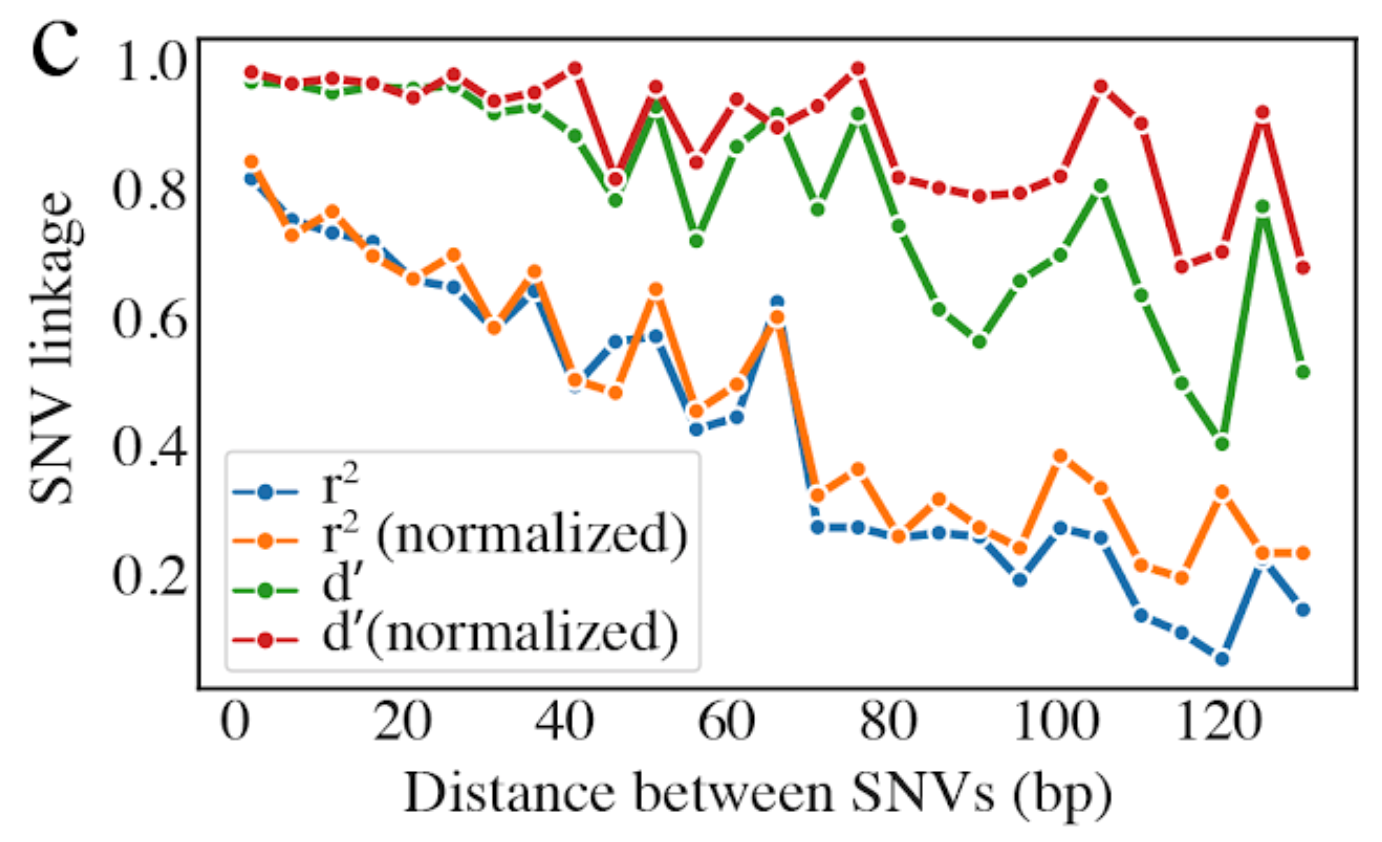
Metrics of SNV linkage vs. distance between SNVs; linkage decay (shown in one plot and not the other) is a common signal of recombination.
6) Read filtering plots¶
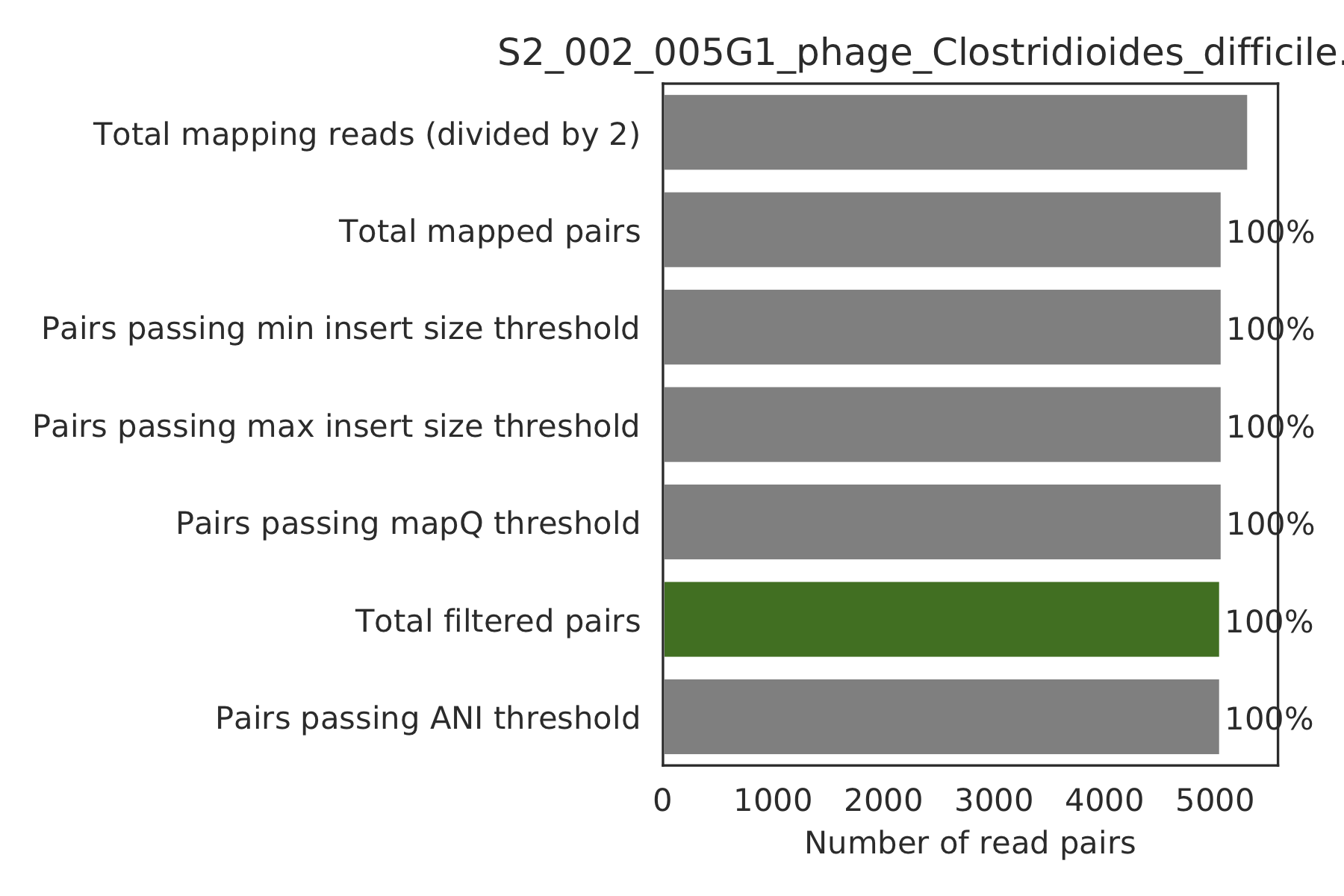
Bar plots showing how many reads got filtered out during filtering. All percentages are based on the number of paired reads; for an idea of how many reads were filtered out for being non-paired, compare the top bar and the second to top bar.
7) Scaffold inspection plot (large)¶
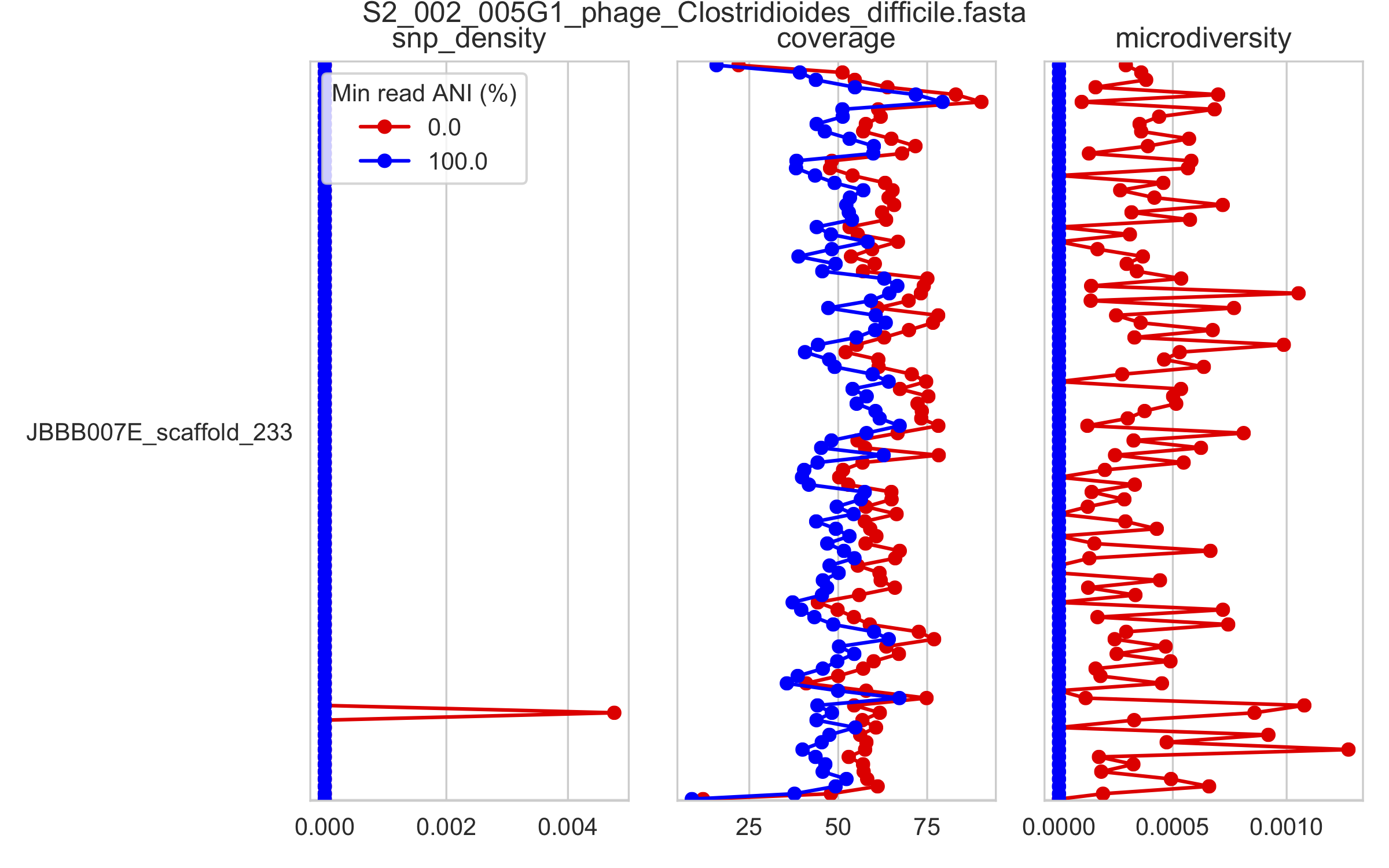
This is an elongated version of the genome-wide microdiversity metrics that is long enough for you to read scaffold names on the y-axis
8) Linkage with SNP type (GENES REQUIRED)¶
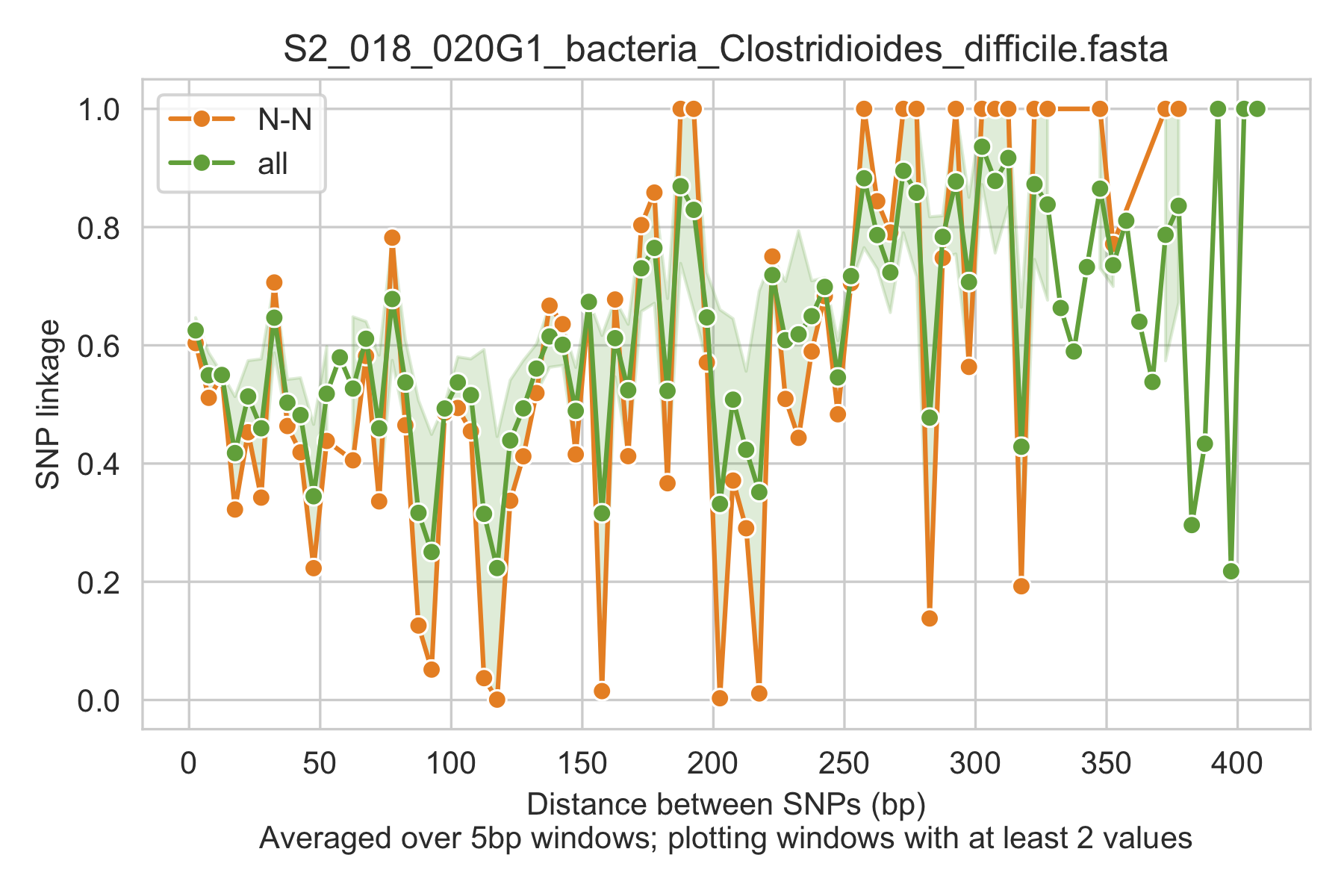
Linkage plot for pairs of non-synonymous SNPs and all pairs of SNPs
10) Compare dendrograms (RUN ON COMPARE; NOT PROFILE)¶
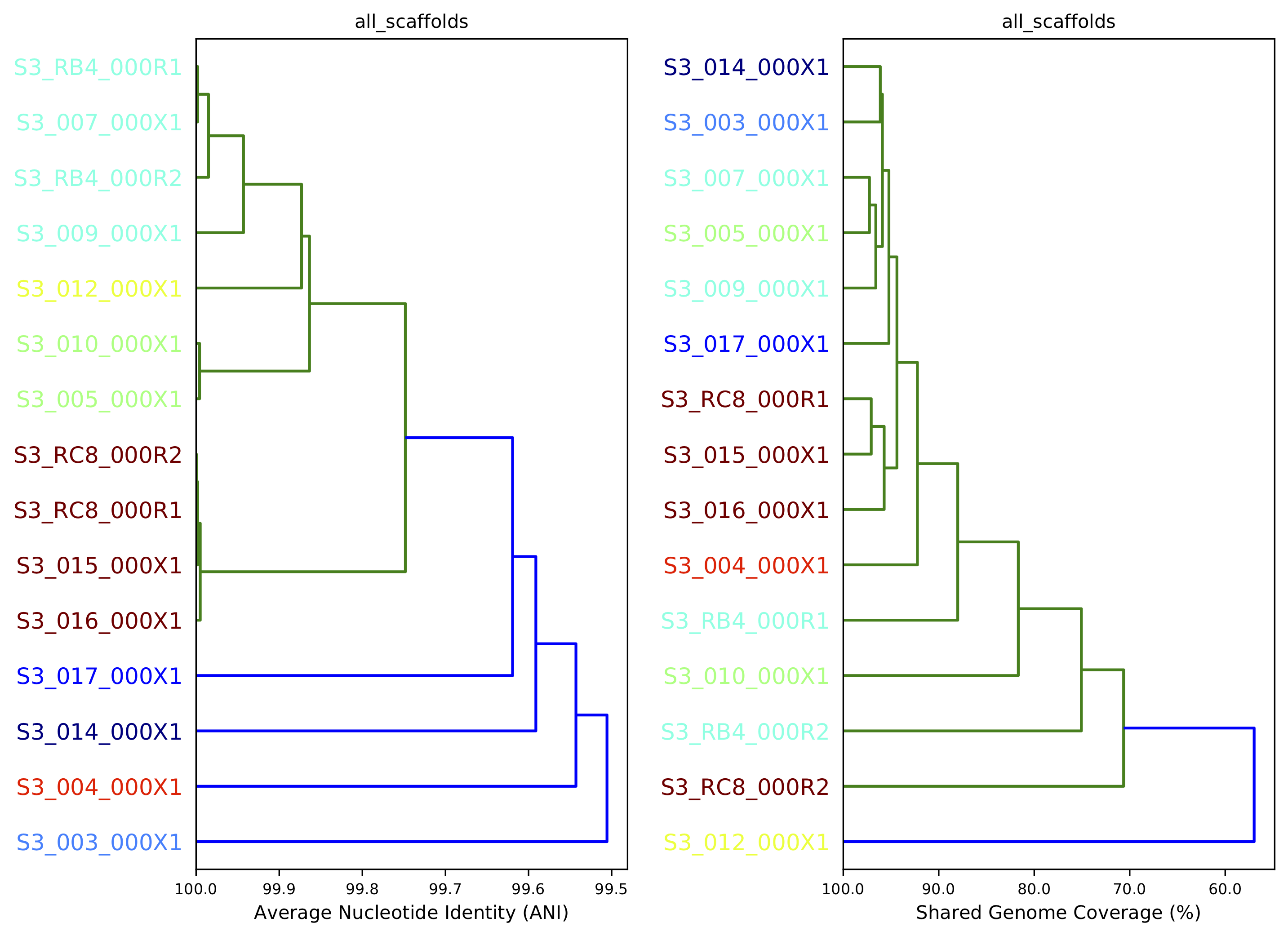
A dendrogram comparing all samples based on popANI and based on shared_bases.